Single Amino Acid Polymorphisms as a Forensically Relevant Source for Personal Identification
Info: 3579 words (14 pages) Dissertation
Published: 9th Dec 2019
Tagged: Forensic Science
Single Amino Acid Polymorphisms as a Forensically Relevant Source for Personal Identification
Abstract
DNA is transcribed, and RNA is formed, the RNA is then translated, and proteins are formed. This is the central dogma of biology. Our DNA is transcribed then translated into proteins, and the proteins are what make up living things. However, identifying these proteins can be a challenge. Nano liquid chromatography (LC) has proven to be “arguably the most common tool for separating proteins” (Wilson et al., 1799), and when coupled with mass spectroscopy (MS) has become a great tool for identifying and modeling proteins. Knowing protein structure, more importantly which amino acids in a protein, has recently become relevant in the field of forensic science as a mode of human identification using polymorphisms in hair proteins. This paper will examine the methods used for protein identification, focusing on LC/MS, and will consider this new area of study as it pertains to forensic science.
Introduction
The central dogma of biology is rather simple. It describes the flow of genetic information (DNA) all the way to proteins, which make up everything from enzymes that break down food, to the structural components of hair. In DNA there are regular random switches in the base pair nucleotides all throughout chromosomes. Figure 1 illustrates the molecular structure of the four nucleotides found in DNA. These random switches also known as single nucleotide polymorphisms (SNPs) are the largest source of genetic variation. They occur mostly in non-coding DNA, however when they are present in coding sequences they can have a range of effects on the phenotype (physical expression of genetic code), such as enzyme function, drug responsiveness, and, most relevant to the scope of this paper, single amino acid polymorphisms (SAPs) (Twyman, 871). Figure 2 demonstrates how a single nucleotide polymorphism can cause an alteration in the amino acid being coded. Understanding the relationship and flow of information from DNA to proteins is a critical aspect involved in protein analysis.
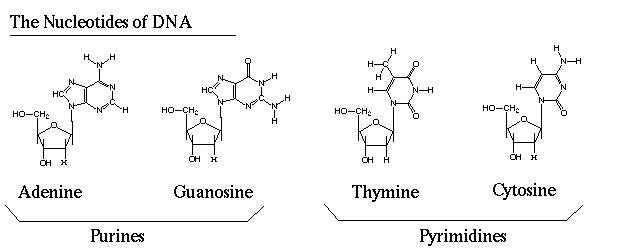
Figure 1: Shows molecular structure of the four nucleotides found in DNA. The purines pair with the pyrimidines; Adenine (A) – Thymine (T) and Guanosine (G) – Cytosine (C) are the pairs that form. This pairing is what causes a single nucleotide switch to cause a change across both strands of DNA. (DNAnucleotides.Gif (620×250))
One of the most vital aspects in analytical science is being able to identify the object being studied. In the field of proteomics there are several methods and tools used for protein identification. Chief among them include Liquid Chromatography accompanied by either Mass spectroscopy or MS/MS. In addition, these techniques are used to identify amino acid sequences, along with databases of protein amino acid sequences are very helpful in locating and identifying SAPs (Xiong et al., 2784).
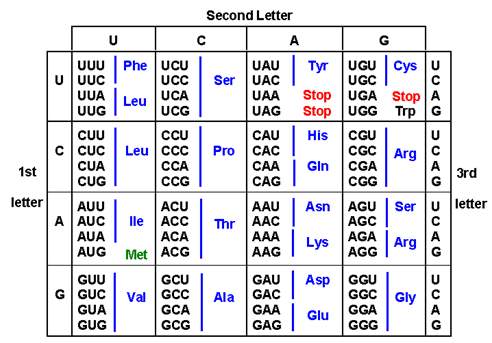
Figure 2: proteins are translated from three nucleotide incremental units. This chart illustrates the possible effects a SNP may have on RNA transcription and in turn alter protein translation in the form of an SAP. (Image from https://qph.fs.quoracdn.net/main-qimg-ccd313266538cb888185b157005c0cdb)
The use of LC-MS has changed the possibilities for the world of protein analysis. Recently a novel study done by Parker et al. observed the possibility of proteins as a source of forensic identification. The study is a response to the problem of DNA degradation. As Parker et al. states “DNA methods depend on the presence of DNA template of sufficient quantity and quality to amplify via PCR and produce genotype information for short-tandem repeat loci (STR), single nucleotide polymorphisms (SNPs), or mitochondrial DNA haplotypes (2). The study observes the use of single amino acid polymorphisms (SAPs) as a source of genetic variation (Parker et al., 1). In addition, not only are proteins a plausible alternative source of genetic variation, proteins are far more stable and abundant than DNA (Parker et al., 2). As previously discussed SNPs inevitably cause SAPs, through transcription then translation, and can be identified using MS.
Hair being such a vital part of many forensic evidence collections, along with its ability to retain substantial amounts of protein, over 300 have been observed, makes it an incredible source of study. According to Parker et al., this amount of protein in hair is enough to “develop forensically and bioarchaeologically useful measures of identity and biogeographic origin” (3).
Experimental
Liquid Chromatography
Chromatography is the separation of some mixture, this is achieved by passing the mixture through a solution or phase. The chemistry of the unique interactions between the components of the mixture and the phase it is suspended in/moving through is what causes the separation. This simplistic version of chromatography explains how liquid chromatography (LC) operates (“Liquid Chromatography”). Specifically, for protein identification and analysis LC is most prevalent. For the purposes of illustrating protein mixture separation this paper will use a theoretical sample mixture of proteins.
To begin the process, it is essential that the sample contains only protein, all precursor steps must be completed to remove any contaminants. A preliminary method of separation may include centrifuging the sample. Further steps to isolate the protein sample will vary and may include “combination of mechanisms such as cell lysis, density gradient centrifugation, fractionation, ultrafiltration, depletion/enrichment, and precipitation” (Tuli and Ressom, 5). According to Tuli and Ressom, cell lysis is done using a variety of methods that include physically disrupting the membranes of cells by shaking with glass beads, or a gentler approach will use a detergent to break the membrane. The purpose of cell lysis is simple, to empty the contents of the cell. Density gradient centrifugation can further help separate after cell lysis, this technique relies on the density of the various components of the lysate to separate during centrifugation. This can be followed by abundant protein depletion or protein enrichment in which proteins of low abundance are targeted. This is done by using specified antibody-based resins which will target the high abundance proteins. Precipitating the proteins in a sample is another regularly used tool, which can be achieved using an organic solvent or salting out. However, using this method requires the precipitating agent to be removed before MS analysis due to ion suppression(Tuli and Ressom, 5).
After isolation, a sample containing an unknown variety of proteins is presented. To sperate and analyze the sample it will be introduced to an LC as illustrated in Figure 3 the sample is pumped through an LC column, this is where separation takes place. The column contains a solid phase, there are two types of columns. According to Tuli and Ressom, reverse phase columns use a polar mobile phase while the stationary phase will be made of a nonpolar material. In normal phase this is reversed, and the stationary phase is polar while the mobile phase used is nonpolar (6). These properties are what allow for the separation, it is because of the affinity of proteins to either the mobile phase being pumped through the LC or the solid phase in the column that creates a unique separation for each protein, which can then be analyzed individually. Due to the nature of proteins reverse phase-LC is best utilized for samples with complex mixtures of proteins and helps encourage analysis by MS (Tuli and Ressom,6). Recent advancements to liquid chromatography have brought the creation of capillary or Nano-LC columns, which instead of using standard sized columns use several smaller columns to help increase accuracy and separation.
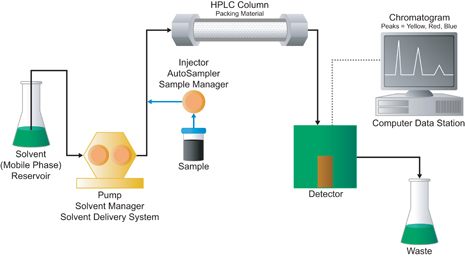
Figure 3: illustrates a basic LC. It depicts the flow of mobile phase, the insertion of a sample, the stationary phase column and finally detection of the separation. (Primer_e_lcsystem.Jpg (465×257))
After separation identification of which proteins are present, and subsequently what the amino acid sequence is accomplished via mass spectrometry (MS).
Mass Spectrometry
“A mass spectrum is a display of the relative abundances of ions produced in an ion source as a function of their mass-to-charge ratios” (Hoffman, 129). Clark, writing in “How the Mass Spectrometer works,” uses a simple analogy of a cannon ball and ping pong ball speeding towards you. To change its direction, a jet of water can be used to hit the balls with perpendicular force to its motion. This would cause the balls to move in a curve trajectory rather than in a straight line. If the same hose and jet of water is used, it is safe to say the force of water would have a greater impact on the forward direction of the ping pong ball. Whereas the cannon ball might move a small amount, the lighter ball would be moved aside far easier. However, each of a definite mass and will be affected by the jet of water uniquely. Clark explains an MS operates very similarly. Molecules in a sample are passed through a stream of electrons, which when enough force is applied may knock an electron off the sample molecule. After being accelerated such that any ions in the sample all have the same kinetic energy, they pass through a magnetic field, which is created via a quadrupole. The sample molecules each act as the cannon ball or ping pong and the magnetic field acts as the jet of water. The degree of deflection is dependent on mass, and detection is dependent on the degree of deflection. Only the ions that make it through the magnetic field are detected, therefore the magnetic field is altered as time passes to allow for different ions to be detected (Clark).
MS/MS, depicted in Figure 4, works very similarly. One of the largest differences between the two is the additional MS. After ions pass through the magnetic field, instead of reaching a detector, they are introduced into a second ionizing chamber, then pass through a second quadrupole. The second ionization is used to selectively isolate an ion with a specific mass. Which is a major asset in protein and amino acid identification and sequencing.
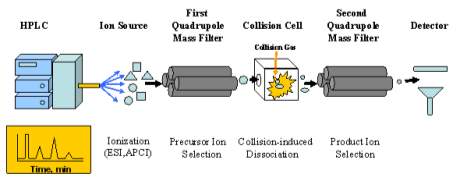
Figure 4: Diagrams an MS/MS, it shows the flow of a sample after it leaves the LC. It is ionized, passes through the first quadrupole, pass through the collision chamber, pass through a second quadrupole, and finally is detected. (Bio_401.Gif (590×268))
In the study conducted by Parker et al. genetic data was produced of the voluntary participants using the Investigative LEAD™ Ancestry DNA Test. This data was used to determine the biogeographic origins of all the individuals using the 190 SNPs identified in the hair proteome, which were “Ancestry Informative Markers” (3). The protein mixtures produced after processing cranial hairs of all subjects, were analyzed using LC-MS/MS. Protein processing included “milling, denaturation, reduction, alkylation, and trypsinization”(Parker et al. 4). The resulting data was then compared to a protein database. After identifying all proteins, a further comparison was done to determine any variants. To insure accurate data collection and analysis, any amino acid variations with a high likelihood of variance occurring after translation, via phosphorylation, were excluded. These amino acid variations included methionine to phenylalanine, asparagine to aspartate, glutamine to glutamate and cysteine to serine (4). In addition to using cranial hair from living subjects, samples were collected from six archeological subjects dating from 1750-1853.
Results
In the study conducted by Parker et al. proteins isolated from cranial hair of study subjects and were identified as having “unique peptide spectral matches” were compared to peptide spectral matching software, to make peptide identifications. Data collected after making matches was compiled into a data base containing single amino acid polymorphisms, which appeared with a > 0.4% frequency. Analysis of peptide sequences resulted in the identification of 89 variant peptides containing 53 single amino acid polymorphisms at 33 genes. To insure accuracy of SAP data analysis, sequencing of 32 allele loci on 22 genes was completed. Results of DNA sequencing indicated 98% positive conclusions while only 2% were falsely identified. Figure 5 illustrates these findings.
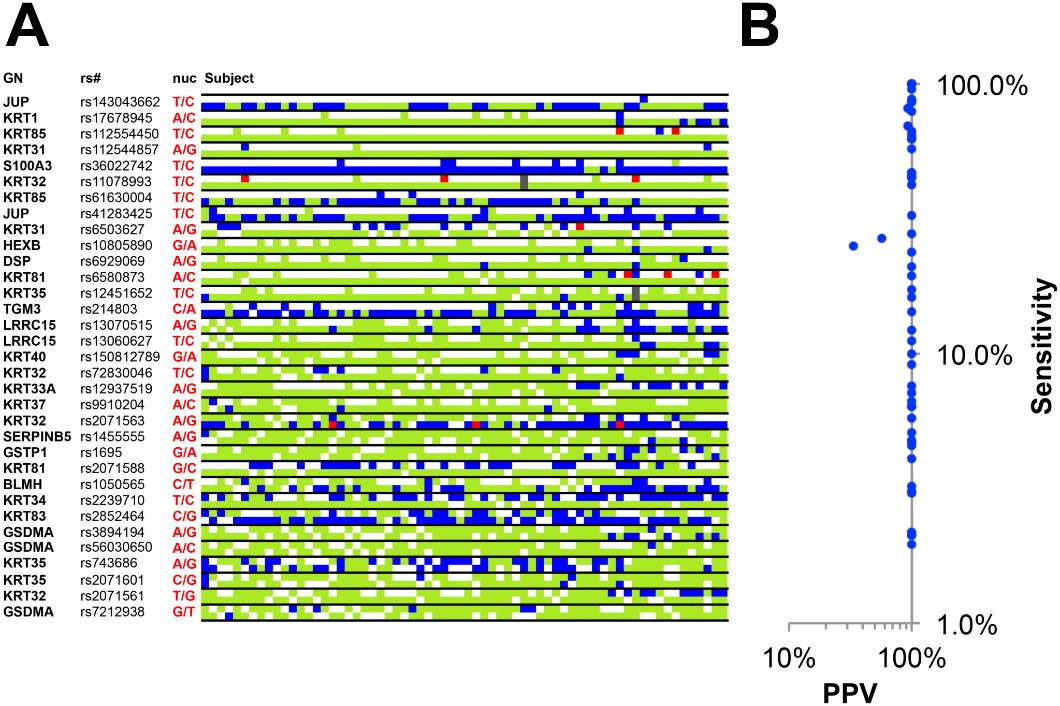
Figure 5: illustrates data collected from Sanger sequencing of DNA in question. Red squares indicate incorrectly predicted single nucleotide polymorphisms (false positives). Blue squares indicate correctly imputed SNPs (true positives). Green squares indicate sequenced alleles which did not have a correlating genetically variant peptide from data analyses (false negatives). Sequencing data absent from both DNA and peptide analyses data (true negatives) are indicated by a white square. While date indicated in grey squares represent failed sequencing determination of SNPs (Parker et al. 6)
Data collected from genetically variant peptides was compared to the current best method of forensic relevant genetic data form hair (mitochondrial DNA analysis). Resulting analysis indicated 60% of genetically variant peptide profiles were “more discriminatory than mitochondrial” DNA analysis (Parker et al. 8).
Samples collected from archeological subjects were digested to isolate the hair proteome and were analyzed via LC-MS/MS resulting protein levels were compared to data collected from living subject MS proteomic spectra. As Parker et al. predicted proteins remaining after this extended period due to environmental processes, were mostly structural in nature. In addition, single amino acid polymorphisms were identified and were associated to single nucleotide polymorphisms(Parker et al. 12).
Discussion and Conclusion
Proteins containing single amino acid polymorphisms have been introduced as a possible source of forensic and archeological identification by Parker et al. Parker et al. states, “SNP alleles can be aggregated to provide a profile of genetic variation for a particular individual (2)” In the study conducted by Parker et al. single amino acid polymorphisms were used to identify single nucleotide polymorphisms in subjects, to test the feasibility of proteins for identification. This data was used to in turn calculate the percentage of other members of the subjects’ biogeographic background having the same single nucleotide polymorphism frequencies. Parker et al. concluded, based on data obtained from both living and archeological subjects, that the probabilities of single nucleotide polymorphisms in a population was comparable to mitochondrial DNA identification methods, as illustrated in Figure 6. According to Parker et al. Mitochondrial DNA is one of the methods currently used for personal identification analysis. Parker et al. further states that, “Current best practice includes sequencing of hair shaft mitochondrial DNA to identify haplotype and sub-clade” (11), explaining the importance of mitochondrial DNA analyses in the face of evidence that is a poor source of nuclear DNA, including hair.
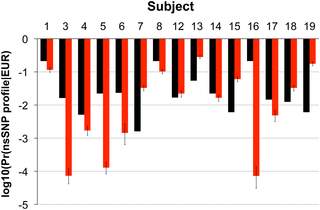
Figure 6: Represents the comparison between the probability of mitochondrial DNA haplotype of a sample from Utah (n=9372) as well as subjects’ from one of the cohorts included in this study (n=15) (represented by the black bars), and the probability of the profiles formed based on this study of single amino acid polymorphisms of the same subjects (n=15) (represented by the red bars). As indicated reliability of single nucleotide polymorphism profiles formed using hair shaft proteins to act as a plausible source of forensically relevance comparable to current mitochondrial DNA use. (Calculations of probability were made using log10). (Parker et al. 9)
According to Parker et al., to insure accurate association of single amino acids polymorphisms to the appropriate single nucleotide polymorphisms, each subjects’ DNA was sequenced and compared to the resulting data obtained from running hair protein samples through LC-MS/MS (13). Data collected and analyzed form subjects indicate a nearly 100% correct prediction of association between single amino acid polymorphisms and single nucleotide polymorphisms, concludes Parker et al. (13). Suggesting that genetically variant peptides can indeed associate “single nucleotide polymorphisms in a subjects’ genome” (Parker et al.13).
The main study described in this paper, conducted by Parker et al. considers the ability of single amino acid polymorphisms for forensically relevant identification, by interpolating these peptide variants to single nucleotide polymorphisms and hence and individuals’ genome. It studies the probability of a certain assigned single nucleotide polymorphism profile appearing in a given population. Although there is promising data in support, Parker et al. conclude that for this approach to become forensically or bioarcheological relevant, further steps towards the development and techniques used must be made (15). In addition, the comparison of single amino acid polymorphisms to mitochondrial DNA is important to this study because it acts as an indication of what is already known and what is yet to come. This study being the first of its kind further highlights the importance of this comparison. Establishing a relevance to the field through comparison, this will further drive research into this area of protein analysis and possible use.
Work Cited
Bio_401.Gif (590×268). http://www.toray-research.co.jp/kinougenri/biology/images/bio_401.gif. Accessed 26 Apr. 2018.
Clark, Jim. “How the Mass Spectrometer Works.” Chemistry LibreTexts, 3 Oct. 2013, https://chem.libretexts.org/Core/Analytical_Chemistry/Instrumental_Analysis/Mass_Spectrometry/How_the_Mass_Spectrometer_Works.
DNAnucleotides.Gif (620×250). http://cs.boisestate.edu/~amit/teaching/342/lab/structure_files/DNAnucleotides.gif. Accessed 30 Apr. 2018.
Hoffman, Edmond. “Tandem Mass Spectrometry: A Primer.” Journal of Mass Spectrometry, vol. 31, 1996, pp. 129–137., doi:10.18411/a-2017-023.
“Liquid Chromatography.” Chemistry LibreTexts, 2 Oct. 2013, https://chem.libretexts.org/Core/Analytical_Chemistry/Instrumental_Analysis/Chromatography/Liquid_Chromatography.
Parker, Glendon J., et al. “Demonstration of Protein-Based Human Identification Using the Hair Shaft Proteome.” PLOS ONE, edited by Francesc Calafell, vol. 11, no. 9, Sept. 2016, p. e0160653. CrossRef, doi:10.1371/journal.pone.0160653.
Primer_e_lcsystem.Jpg (465×257). http://www.waters.com/webassets/cms/category/media/other_images/primer_e_lcsystem.jpg. Accessed 23 Apr. 2018.
Tuli, Leepika, and Habtom W. Ressom. “LC–MS Based Detection of Differential Protein Expression.” Journal of Proteomics & Bioinformatics, vol. 2, Oct. 2009, pp. 416–38. PubMed Central, doi:10.4172/jpb.1000102.
Twyman, R. M. “Single-Nucleotide Polymorphism (SNP) Analysis.” Encyclopedia of Neuroscience, edited by Larry R. Squire, Academic Press, 2009, pp. 871–75. ScienceDirect, doi:10.1016/B978-008045046-9.00866-4.
Wilson, Steven Ray, et al. “Nano-LC in Proteomics: Recent Advances and Approaches.” Bioanalysis, vol. 7, no. 14, Aug. 2015, pp. 1799–815. Crossref, doi:10.4155/bio.15.92.
Xiong, Yun, et al. “An NGS-Independent Strategy for Proteome-Wide Identification of Single Amino Acid Polymorphisms by Mass Spectrometry.” Analytical Chemistry, vol. 88, no. 5, Mar. 2016, pp. 2784–91. Crossref, doi:10.1021/acs.analchem.5b04417.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Forensic Science"
Forensic science, or forensics, is the application of science to criminal and civil law, usually during criminal investigation, and involves examining trace material evidence to establish how events occurred. Forensic scientists provide impartial scientific evidence that can be used in court.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: