Harnessing Albumin as a Carrier for Cancer Therapies
Info: 15840 words (63 pages) Dissertation
Published: 16th Dec 2019
Tagged: Cancer
Harnessing Albumin as a Carrier for Cancer Therapies
- Introduction
- Albumin as a Carrier
- Cancer Implications of Albumin
- Passive Albumin Tumor Accumulation
- Tumor Albumin Metabolism
- General Albumin Binding Strategies
2.1 In Situ Binders
2.1.1 Covalent Conjugation
2.1.2 Native Ligand Conjugates
2.1.3 Small Molecule Binders
2.1.4 Albumin Binding Domain and Nanobodies
2.2 Exogenous Formulations
2.2.1 Covalent Conjugation
2.2.2 Nanoparticle
2.2.3 Recombinant Albumin Fusion Proteins
- Survey of Results
3.1 Chemotherapeutics
3.1.1 Nanoparticles
3.1.2 Covalent Conjugates
3.2 Biologics
3.2.1 Oligonucleotides
3.2.2 Immunomodulatory Drugs
3.3 Theranostics
3.4 Clinical Results
- Conclusions and Future Directions
Acknowledgements
References
Abstract
Serum albumin, a natural ligand carrier that is highly concentrated and long-circulating in the blood, has shown remarkable promise as a carrier for anti-cancer agents. Albumin is able to prolong the circulation half-life of otherwise rapidly cleared drugs and, importantly, promote their accumulation within tumors. The applications for using albumin as a cancer drug carrier are broad and include both traditional cancer chemotherapeutics and new classes of biologics. Strategies for leveraging albumin for drug delivery can be classified broadly in to exogenous and in situ binding formulations that utilize covalent attachment, non-covalent association, or encapsulation in albumin-based nanoparticles. These methods have shown remarkable preclinical and clinical successes that are examined in this review.
1. Introduction
Albumin, a long-circulating and highly-abundant protein in the blood, has unique promise as a carrier for cancer therapeutics based on several key characteristics: (1) it is a natural carrier of native ligands and other hydrophobic cargo (2) it is rescued from systemic clearance and degradation by natural mechanisms (3) it accumulates at sites of vascular leakiness and (4) it is more highly taken up and metabolized by rapidly growing, nutrient-starved cancer cells. Investigators have sought to leverage these characteristics for the delivery of several classes of approved and investigational anticancer agents, which will be reviewed herein.
1.1 Albumin as a Carrier
Albumin is the most abundant protein in human blood with a concentration of about 40 mg/mL and a molecular weight of ~67 kDa [1]. Notably, it exhibits an extraordinarily long half-life of 19 days [2] [3]. Albumin is synthesized in the liver with approximately 13-14g of albumin entering the circulation every day [2]. When albumin extravasates into the tissue, it is returned to the vascular space via the lymphatic system through a nature recycling mechanism. The same approximate mass of 13-14g of albumin entering the intravascular space is also catabolized from it every day. Importantly, albumin is known to be a carrier of a wide variety of both endogenous and exogenous compounds [4]. This facilitates the colloidal solubilization and transport of normally hydrophobic molecules such as long chain fatty acids as well as a variety of other ligands such as bilirubin, metal ions such as zinc and copper, and therapeutic drugs such as warfarin and ibuprofen [5]. Figure 1shows the crystal structure of albumin and the sites where these ligands can bind. Some albumin-bound ligands may also benefit from albumin shielding the cargo from degradative serum enzymes.
Albumin naturally transcytoses across the vascular endothelium. This process is attributed to the receptor GP 60, also known as albondin. GP 60 is a receptor present in continuous vascular endothelium and alveolar epithelium. Albumin binds to GP60 which induces clustering of albumin-gp60 at cell surface and association with Cav-1, the main protein critical to caveolae formation. Cav-1 induces invagination of surface membrane surrounding the clustered GP 60-albumin receptors and the subsequent internalization of a vesicle composed of albumin and compounds bound to albumin. Cav-1 is transported to and fuses with basolateral membrane and completes transcytosis. [2] (Figure 2A) However, GP 60 only binds to native albumin. GP 18 and GP 30, by comparison, exhibit preferential binding to modified albumin to traffic it for lysosomal degradation and are widely distributed in the body [7]. These receptors may be part of a pathway to remove old, damaged, or potentially deleterious albumin [8]. Albumin-gold conjugates and maleic anhydride treated albumin, for example, are modified forms of this protein subject to this pathway, necessitating careful design consideration for the modification of albumin for drug delivery [7].
The half-life of albumin is one of its remarkable properties that make it an attractive carrier for improving the pharmacokinetic properties of anticancer agents. This long half-life is attributed to neonatal Fc receptor (FcRn), an intracellular receptor that is responsible for rescuing albumin from degradation. This receptor is widely distributed in the body and is known to extend the half-life of both serum albumin and immunoglobulin G. The FcRn functions by binding these proteins in the acidic endosome and diverting them from the highly-degradative lysosomal pathway. Both molecules are exocytosed into the extracellular compartment where, at physiological pH, they are released from the FcRn. Albumin can then re-enter the circulation through the lymphatics, thereby prolonging its half-life. [2] This receptor interaction is pH dependent with strong affinity binding occurring at the low pH of the endosome (Figure 2B). Additionally, albumin can avoid renal clearance by reabsorption through the megalin/cubilin receptors. Cubilin is a 460 kDa glycoprotein which binds to transmembrane endocytic receptor megalin and has been demonstrated to be important for binding albumin and tubular albumin reabsorption [9]. Albumin binds cubilin with high affinity (Kd~0.6µM), and studies have shown that megalin facilitates the endocytosis and intracellular trafficking of cubilin [10]. Both megalin and cubilin are highly expressed in the renal proximal tubule brush-border and endocytic apparatus, and it is thought that megalin may be directly involved in reabsorption as an albumin receptor, and/or indirectly by affecting the expression and/or endocytic function of cubilin [10]. Moreover, as a result of the size selective properties of the glomerular filter, the primary urine contains proteins of low molecular weight (<60 kDa), whereas larger proteins are excluded [11]. Using albumin as a carrier can leverage this natural pathway for extending drug circulation half-life. This approach circumvents the usage of synthetic systems which may involve complicated synthesis and confer some toxicity or immunogenicity. For instance, modifying drugs with poly(ethylene glycol) is a common method for improving hydrophilicity and circulation of a drug by making it larger the renal size cutoff. However, humans can mount antibody responses to PEG, resulting in potentially dangerous allergic reactions and potentially limiting the utility of repeated administration of PEG-containing formulations [12]. The biocompatibility and physiological transport pathways used by albumin make it an exciting platform for anticancer drug development.
- Cancer Implications of Albumin
1.2.1 Passive Albumin Tumor Accumulation
The internal vascular network of growing tumors often becomes insufficient for supplying the oxygen and nutrients necessary to support their aberrant, hyper-proliferative local environments. Consequent hypoxic conditions and cell death are associated with the release of angiogenic factors that cause rapid formation of new blood vessels at the tumor site. These new vessels are characterized by irregularities such as larger than normal fenestrations in the endothelium [13]. Tumors also have disruption of the lymphatic system, which in healthy tissues, continuously drains the extracellular fluid, facilitating reentry of macromolecules into the circulation. In tumors, there is either poor or heterogeneous lymphatic drainage due to the compression and eventual collapse of lymphatic vessels by rapidly growing cancer cells [13][14]. The tumor vasculature leakiness, paired with poorly formed lymphatic drainage, is thought to be responsible for the preferential tumor accumulation of nanostructures and macromolecules in the phenomenon known as the enhanced permeability and retention effect (EPR) (Figure 2C) [15].
Albumin is especially adept at accumulating in regions of proliferating tumor cells as a consequence of the EPR effect. This is because albumin is the most concentrated protein in the blood at the aforementioned concentration of 40 mg/mL compared to its interstitial concentration of about 14 mg/mL, driving its diffusional transport [16]. Furthermore, the molecular weight of albumin (67 kDa) is near the reported optimum size of ~50 nm nanomedicine for deep tissue penetration and high retention in tumors [17]. Additionally, the reliance of albumin on the lymphatic system to return to the circulation from the extracellular space makes it susceptible to accumulation in tumors with their poor lymphatic drainage[15] [18]. Indeed, the first observations of macromolecule accumulation in tumor interstitium were based on the preferential distribution and retention of radiolabeled albumin and other serum proteins [15]. Importantly, Evans blue dye, which naturally complexes with albumin, demonstrates prolonged retention in tumors compared to normal tissue, from which it is rapidly cleared [15] . Albumin accumulation has since been observed in a variety of solid tumors animal models including sarcoma, ovarian carcinoma, and Novikof hepatoma [19].
The EPR effect has recently come under scrutiny based on the disparity observed between nanocarrier tumor biodistribution and therapeutic efficacy in preclinical tumor models versus human clinical trials. Tumor vasculature formation occurs at a faster rate in commonly used flank tumor mouse models compared to most human disease, and this physiologic difference may exaggerate the EPR effect in some animal models. Additionally, there is growing appreciation that the EPR effect may be more relevant for certain tumor or patient subsets among the wildly heterogeneous spectrum of human cancers. The development of companion diagnostic nanoparticles is a promising approach for predicting patient responses to nanomedicine, which may be key to best leveraging the EPR effect for clinical delivery [20]. Interestingly, in observations among a variety of tumor models including syngeneic breast tumors, mouse mammary intraepithelial neoplasia outgrowths, and epithelial-mesenchymal transition tumors, permeability to albumin is ~4 fold greater than 100 nm liposomes [21]. Further, the prolonged circulation time associated with albumin may further enhance tumor accumulation by EPR. These finding suggest promise for the improved penetration of albumin therapeutics.
1.2.2 Albumin Tumor Metabolism
In addition to the desirable passive tumor tissue accumulation of albumin, it also demonstrates preferential uptake by tumor cells which can be leveraged for intracellular delivery of therapeutic cargo. In 2013, Commisso et al observed a mechanism by which cancer cells can support their increased metabolic and growth needs by active uptake of extracellular proteins through micropinocytosis. They observed that cancer cells expressing oncogenic Ras, an inner plasma membrane protein whose aberrant activation is associated with virtually all aspects of the malignant cancer phenotype, more highly utilize extracellular proteins as a source of amino acids to drive cellular growth [22][23]. Indeed, pancreatic ductal adenocarcinoma cells have since been shown to grow indefinitely in media lacking essential amino acids in the presence of physiologic albumin [24]. These findings provide important insight into the ability of albumin to be internalized by cancer cells in a more targeted manner. It has been additionally observed that hypoalbuminemia is a characteristic feature of patients with advanced solid tumors [25]. Decreased serum albumin in these patients may be indicative of the increased catabolism of albumin by the proliferating tumor as an abnormal source of amino acids utilized to meet high metabolic demands [26].
Some cancer cells also preferentially use receptor-mediated albumin uptake pathways in addition to upregulated use of nonspecific macropinocytic mechanisms. This has become particularly evident based on the discovery that a correlation exists between expression of albumin-related receptors and relative efficacy of albumin-based therapies among different cancer types. Nab-paclitaxel (nab-P), also known as Abraxane, is an FDA approved albumin-bound paclitaxel particle. Recently, Chatterjee et al attempted to elucidate why certain pancreatic cancer patient populations responded better to treatment with nab-P than others [27]. It had previously been posited that SPARC (secreted protein acidic and rich in cysteine) was a critical albumin-binding protein that facilitates the efficacy of nab-P in metastatic pancreatic cancer. This idea was based on small scale retrospective studies (n=16) on nab-P treatment of head and neck cancer where the relationship was examined between SPARC tumor expression and patient outcomes [28]. This hypothesis was centered around the notion that the presence of SPARC in the tumor environment would concentrate nab-P and thus possibly enhance its therapeutic effect. However, further exploration in a phase III clinical trial on nab-P in metastatic pancreatic cancer patients demonstrated no association between SPARC level and treatment efficacy [29]. Indeed, additional preclinical studies in a mouse model of pancreatic cancer treated with nab-paclitaxel plus gemcitabine also demonstrated a lack of association with SPARC knockout and tumor progression [29]. Chatterjee et al instead examined the role that caveolae, omega-shaped invaginations of the plasma membrane, play in albumin uptake in cancer cells [30]. Caveolae have previously been implicated in a wide variety of cellular processes including endocytosis, transcytosis, and signal transmission [31][32]. The primary protein necessary for caveolae formation, Caveolin-1 (Cav-1), is upregulated in a wide variety of cancer types including pancreatic cancer, prostate cancer, and breast cancer and is associated with cancer progression [27], [33]–[35]. It has been shown in Cav-1 knockdown mice that caveolae are critical in albumin uptake and transport [36]. Interestingly, Chatterjee et al demonstrated that Cav-1 expression is critical to the entry and tumor response of nab-P in pancreatic cancer models. This new development in the mechanistic understanding of albumin-bound chemotherapeutics may aid in better identification of patients who are likely to respond to albumin-based therapies based on prescreening of tumor biopsies. By exploiting the natural affinity of albumin for tumor biodistribution and preferential tumor cell uptake, albumin provides great promise as a carrier for increasing the therapeutic efficacy of cancer drugs.
2. General Albumin Binding Strategies
Multiple classes of albumin-based formulations have been tested for cancer-targeting therapies (Figure 3). Herein we will review two general categories- preformed albumin therapeutics and in situ binders. In situ binders can dock on to endogenous albumin after delivery into the body. Exogenous formulations, by comparison, rely on drug loading in or attachment to recombinantly produced albumin, bovine serum albumin, or human serum albumin isolated from donors prior to administration to patients. This discussion will focus specifically surveying albumin-based cancer therapeutics, but other types of clinically-mature therapeutics will be highlighted in each class as applicable.
2.1 In Situ Binders
2.1.1 Covalent Conjugation
Covalent bonds between native albuminand therapeutics can be formed in situ. The primary method for in situ covalent attachment to albumin leverages the cysteine-34 amino acid of albumin. Importantly, albumin cysteine-34 represents the most abundant free thiol in the blood, and competitive side reactions with other free thiols are not a significant concern because cysteines are typically found in nonreactive disulfide bridges. Kratz and colleagues pioneered this method for binding endogenous albumin [37]. Their approach used a maleimide carboxylic hydrazone derivative of doxorubicin to form a covalent thioether bond in situ with the cysteine-34 position of albumin. This approach is made possible by the unique properties of this amino acid residue, with approximately 70% of circulating albumin possessing the Cys-34 amino acid in its accessible form. Indeed, the authors posit that the lack of a free thiol on the majority of other circulating serum proteins makes this a relatively specific reaction. The preferential reaction with albumin is further bolstered by the low cysteine 34 pKa of 7, making it the most reactive thiol group in human plasma. The incorporation of an acid-sensitive hydrazone linker was utilized by the authors to create triggered release of the doxorubicin cargo within the highly acidic environment of endosomes and lysosomes. A variety of groups have used this approach to improve the pharmacokinetic properties of their cancer therapeutics ranging from small molecule drugs [38]–[42] to biologics [43]. Additionally, rather than employing the maleimide to sulfhydryl reaction, a disulfide bond with this free thiol can be formed to make a more readily reducible link between therapeutic cargo and native albumin. One such example involves the addition of a cysteine residue to a tumor penetrating peptide to form a disulfide bond with albumin in situ [44]. Finally, both ruthenium-based anticancer complexes and copper pro-drugs have been synthesized to bind endogenously to the large hydrophobic cavity at the IIA subdomain of albumin, followed by subsequent exchange with the N-donor residues of Lys 199 and His 242 to form a stable albumin complex [45], [46].
Several strategies have been employed to facilitate the liberation of the therapeutic cargo from albumin after it reaches the site of interest. In addition to the acid-labile hydrazone and reducible disulfide linkages mentioned above, another example albumin-binding prodrug incorporated a caspase cleavable peptide spacer [47] to promote drug release from albumin at sites of tumor radiotherapy. In this design, the DEVD peptide, a well-known substrate of caspase- 3, was attached on one end to albumin cysteine-34 through a maleimide functional group and on the other end to doxorubicin by a self-immolative linker. Because the enzyme caspase-3 is upregulated during apoptotic cell death that occurs in an irradiated tumor, this design allows for targeted release of doxorubicin chemotherapy at the site of tumor radiotherapy.
An alternative liberation approach from in situ covalently bound albumin involves cathepsin cleavable linkers. Cathepsins B and D are lysosomal enzymes known to be overexpressed in a variety of malignant tumors [48]. In their prodrug, Schmid and colleagues incorporated a pentapetide linker, Ala-Leu-Ala-Leu-Ala, which is a known cathepsin cleavable sequence previously employed in targeted drug release [48]. The group conjugated ε-maleimidocaproic acid to camptothecin (a small molecule inducer of apoptosis) and doxorubicin using this linker flanked by two arginines which were incorporated to promote water solubility. A final method for liberating cancer therapeutics covalently bound to endogenous albumin leverages MMP-2 linkers. MMP-2, a matrix metalloproteinase, has been shown to be overexpressed in melanoma and plays a significant role in tumor proliferation, angiogenesis, and metastasis [49]. Mansour and colleagues introduced an octapeptide, Gly-Pro-Leu-Gly-Ile-Ala-Gly-Gln, that has been shown to be effectively cleaved by MMPs 2 and 9 when used to link doxorubicin and an albumin-binding maleimide group [49]. These covalent binding and targeted release strategies could be generalized for the delivery of a wide range of cargo including other small molecule drugs and biologics.
2.1.2 Native Ligand Conjugates
Another approach to promote in situ interaction with albumin to alter drug pharmacokinetics involves the conjugation of therapeutic agents to ligands that naturally bind albumin non-covalently in the body. For instance, fatty acids are naturally ferried by albumin in the bloodstream due to their insolubility in plasma. Indeed, albumin is known to have seven fatty binding sites distributed asymmetrically on the protein and to bind around ~0.1 mol of fatty acid per mol human serum albumin under normal physiological conditions [19]. A common approach to promoting in situ docking of therapeutics through the fatty acid binding pockets of albumin involves direct conjugation of fatty acids to therapeutic payloads. Notably, commercially available Semaglutide (Figure 3) uses an 18-carbon fatty diacid linked to a glucagon-like peptide-1 analog via a glutamyl ethylene glycol spacer for the treatment of diabetes albumin binding Kd~0.38 nM and half-life of 46.1 hr in mini-pigs) [50]. This was a remarkable improvement compared to its precursor, Liraglutide, (16-carbon monoacid with γGlu spacer) which demonstrated an i.v. half-life of 8-10 hours. This drastic enhancement underscores the impact of both linker and binding chemistry in the use of albumin as a drug carrier. Similar noteworthy approaches have used a diacyl lipid linked by polytheylene glycol to deliver antigen/adjuvant cargo [51], palmitoyl modifications to the 2’ position of antisense oligonucleotides [52], and DNA nanocages modified with branching 12 carbon chains for encapsulation of cargo[53]. Interestingly, increasing the valency of lipid-modified drugs appears to result in superior albumin-binding and pharmacokinetics, but may hinder potency depending on modification site [54].
In the context of cancer treatment, Sarrett et al used lipid-mediated in situ albumin binding to deliver therapeutic short interfering RNA (siRNA) to solid breast tumors. Short interfering RNA is a powerful gene silencing platform that involves the degradation of a specific mRNA, which can be used to directly target expression and phenotypes associated with a wide spectrum of cancers. However, siRNA pharmacokinetics are typically poor because it is subject to rapid renal clearance when introduced in the body. Further, unmodified siRNA has no mechanism for targeting cancer cells or reaching their cytosolic site of action. This approach involved using simple and specific “click” chemistry to conjugate modified siRNA to a PEGylated diacyl lipid, and resulted in a 5.7-fold increase in half-life [55]. Similarly, cholesterol is another native ligand that is non covalently bound to albumin in the body and has been used as a means to improve the pharmacokinetic properties of biologics [56] [57], [58]. In a study on a variety of lipophilic siRNA conjugates including cholesterol, oleyl alcohol, lithocholic acid, and oleylamide of lithocholic acid modifications, it was found that a 6 to 10 carbon aliphatic linker conferred the optimal uptake and gene silencing properties; shortening of the linker reduced efficiency of cellular uptake, whereas lengthening increased uptake but decreasing silencing efficiency in KB-8-5, HepG2, and HEK 293 cells [59]. These lipophilic moieties allow for non-covalent association of cargo which may circumvent hindered pharmacokinetic properties and allow for easier offloading into sites of interest due to reversibility.
2.1.3 Small Molecule Binders
In contrast to leveraging native ligands, synthetically produced small molecules can also be used for binding to endogenous albumin. A notable example of these small molecule binders is the dye Evans Blue (EB). Remarkably, each albumin protein can bind up to 14 Evans Blue molecules [60]. Truncated versions of this molecule have been derived to retain its albumin-binding properties but allow for modifications such as conjugation to other drugs or imaging agents[61] [62][63]. For instance, truncated EB conjugated to the anti-diabetic drug Exendin-3 resulted in markedly improved half-life (5 to 32 hours) and yielded improvement in hypoglycemic effects [64] [65]. A recent and interesting use of this platform was the development of an albumin/vaccine nanocomplex that conjugated molecular vaccines to EB derivatives for use in cancer immunity (Kd for mouse serum albumin = 1.0 µM )[66].
Another small synthetic small molecule binder was developed by Dumelin and colleagues as a “portable” albumin binder using a DNA-encoded chemical library [67]. The resulting binders were characterized by a common butanoyl moiety and require hydrophobic groups in the para position of the phenyl ring for good retention. The most successful candidate was a 4-(p-iodophenyl)butyric acid derivative that demonstrated affinity for albumin with Kd=3.2µM. Conjugation with this low molecular weight albumin binding entity has prolonged half-life of antibody fragments (~30 mins to 1000 mins) [68] and reduced renal clearance of a chelator-folate conjugate (∼70 %ID/g vs 28 %ID/g at 4 h after injection) [69]. Other groups have found that a wide variety of synthetic and naturally occurring aromatic compounds can be used for non-covalent association with albumin, which may be attributed to their hydrophobic nature. For instance, the FDA-approved MRI agent gadofosveset trisodium utilizes a 4-diphenylccyclohexyl group to reversibly bind serum albumin, resulting in extension of elimination half-life from approximately 90 to 1000 minutes [70] [71]. Modifications with these simple albumin-binding moieties are a synthetically appealing approach for improving drug pharmacokinetic properties.
2.1.4 Albumin-Binding Domains and Nanobodies
Albumin-binding peptides have also been leveraged to non-covalently associate with albumin to improve the pharmacokinetics of cancer therapeutics. These albumin-binding domains (ABDs) are often genetically fused to recombinantly-produced therapeutic proteins. The heptapeptide WQRPSSW is an albumin-binding domain that was identified using phage display by Su and colleagues [72]. This group genetically fused the chimeric peptide BB28, a fusion of tumor homing bombesin and mitochondria disrupting peptide, to this albumin binding domain in order to extend its half-life from several minutes to 2 hours [72]. Li et al, by comparison, used a 46-amino acid albumin-binding domain derived from streptococcal protein G [73] [74]. Streptococcal protein G is a cell-surface exposed protein produced by gram-positive bacteria that is capable of binding to serum proteins, namely albumin [75]. This group sought to extend the half-life of human recombinant tumor necrosis factor-related apoptosis-inducing ligand (hTRAIL), a tumor specific inducer of apoptosis whose lack of efficacy in clinical trials has been attributed to its poor pharmacokinetic properties. Their study identified fusion of the N terminus of this 46-amino acid ABD to hTRAIL as a promising technique for improving its antitumor efficacy. The resulting conjugate bound human serum albumin with high affinity (Kd=0.4nM) and extended the half-life of hTRAIL from 0.32h to about 14.1h. Other successful examples of this technique include ABD genetic fusion to human soluble complement receptor type 1 [76], insulin-like growth factor II [77], and respiratory syncytial virus subgroup protein [78].
Monoclonal antibodies are a common means for high affinity binding to biologic targets and have had marked success in clinical translation. However, intact IgG antibodies are large (~150 kDa), and thus are considered to have limited tumor penetration capability [79][80]. This challenge has prompted investigation into monoclonal antibody fragments to achieve high affinity target binding with a molecule that is smaller in size. However, these antibody fragments can have diminished target affinity and can be too small in size to avoid renal clearance. To overcome these obstacles, Tijink et al investigated the class known as nanobodies for binding to albumin [79]. Nanobodies are derived from the unique antibody format present in dromedaries that is comprised only of the heavy chain. These “heavy chain only” antibodies are approximately 15 kDa, but, upon the desired binding to albumin would be able to avoid renal clearance. Tijink and colleagues constructed a 50 kDa albumin-binding nanobody for the purpose of improving upon existing epidermal growth factor receptor (EGFR) antibody treatments. Their bivalent nanobody comprised two EGFR binding units and one albumin binding unit [79]. The group details that, by themselves, nanobodies are quickly excreted, making intermediate association with albumin an additionally appealing approach. The EGFR-EGFR-Alb nanobody showed faster and deeper tumor penetration than the unmodified EGFR-EGFR binder in A431 xenograft-bearing mice with a tumor to blood ratio greater than 80 achieved after 6 hours.
Another interesting implementation of albumin-binding domains involves its fusion to an affibody molecule. Affibodies are a new class of small (~6.5 kDa) proteins with high target specificity derived from staphylococcal protein A [81].Multiple groups have sought to fuse an albumin binding domain to affibody targeting human epidermal growth factor receptors, which are overexpressed in a variety of cancers and are known to drive tumor cell proliferation [82] [83]. Interestingly, Orlova and colleagues were able to incorporate a radiolabel into these affibody molecules and suggest the potential of labeling with therapeutic radionuclides for radioimmunotherapy of solid tumors [82]. The elimination half-life of the parent affibody after fusion to the ABD was increased 80-fold from 0.5 to 41h. Similarly, a 46-amino acid ABD derived from streptococcal protein G was used to create a bispecific single-chain diabody for the retargeting of cyototoxic T cells to carcinoembryonic antigen (CEA)-expressing tumor cells by genetic fusion. This diabody was directed against the CEA and T cell receptor complex protein CD, and the resulting complex resulted in a 6 fold circulation time extension [84].
2.2 Exogenous Formulations
2.2.1 Covalent Conjugation
Cancer drug formulation with exogenous albumin prior to delivery into the body has also been pursued in several formats, including direct, covalent conjugation of drug to albumin. A common strategy is to conjugate therapeutic cargo to a primary amine available within a free lysine residue on albumin. This strategy has been used for the conjugation of small molecule drugs such as methotrexate [85], curcumin [86], and doxorubicin [87]. These albumin-drug conjugates used the formation of amide bonds and reductive amination for covalent attachment. The benefits to this covalent method include avoiding use of albumin’s free thiol which is not always available, and the presence of multiple lysine groups available during exogenous conjugation. However, Kuhlmann et al suggest that, although this method allows for the conjugation to multiple lysines, the absence of selectivity may compromise FcRn engagement and consequent albumin pharmacokinetics. Additionally, it may be more difficult to control the number of modifications per albumin as well as site specificity. The aforementioned binding method for conjugating drugs to the cysteine-34 of albumin can also be used for covalent binding to exogenous human serum albumin. Indeed, this method was used for the conjugation of oligodeoxynucleotides to this position to allow for the subsequent annealing of various complementary strands such as aptamers [88]. However, these exogenous albumin binding methods requires either donors or recombinant production, both of which are associated with their own challenges including possible transmission of disease and high cost.
2.2.2 Recombinant Albumin Fusion Proteins
Direct genetic fusion of therapeutic proteins to whole recombinant albumin is another alternative. For instance, this approach was used to link the N-terminus of proaerolysin, a potent toxin, to recombinant albumin [89]. This group has termed the resulting therapeutic a “pro-toxin”. The goal is for these “pro-toxins” to only be cleaved by a defined protease that is present in the metastatic prostate cancer tumor microenvironment. The authors specifically engineered a peptide linker into their recombinant protein that was specific for the protease prostate specific antigen. Another recombinant HSA fusion protein, Albuleukin, combines recombinant interleukin-2 (rIL-2) and human serum albumin. This strategy aimed to maintain the excellent pharmacokinetic properties of albumin while conferring the immunomodulatory and anti-tumor properties of rIL-2 [90]. Other examples of this strategy include an interferon-β [91], anticarcinoembryonic antigen single-chain antibody [92], and barbourin [93]. This method is elegant in the sense that it does not require any complicated conjugation chemistry, merely the isolation of precisely-defined, recombinant protein.
2.2.3 Nanoparticle Formulations
One of the more widely explored methods that utilize albumin as a carrier for cancer therapeutics involves drug encapsulation into an exogenous albumin-based nanoparticle. The appeal of this method lies in the ability to leverage the native albumin mechanisms that facilitate its extraordinarily long half-life and cancer homing properties, while shielding therapeutic cargo until the particle is broken down at the therapeutic site of interest. The methods for synthesizing albumin nanoparticles can be generally categorized into the techniques of desolvation, emulsification, thermal gelation, and more recently, nano spray drying, and self-assembly [94] (Table 1). The most notable of these albumin nanoparticles is the aforementioned FDA approved nab-paclitaxel or Abraxane. Nab-paclitaxel synthesis involves passing a lipophilic drug and human serum albumin in an aqueous solvent through a jet under high pressure to form nab-paclitaxel nanoparticles with a mean particle size of 130 nm [95][96]. However, various albumin nanoparticle strategies have been employed for a wide variety of treatment agents and a myriad of chemical modifications. Herein, we will survey notable examples of the large pool of clinical and preclinical formulations that utilize the various aforementioned synthesis methods.
| FabricationMethod | Description | RepresentativeExamples |
| Desolvation (Coacervation) | Desolving agent such as ethanol or acetone is continuously added to an aqueous solution of albumin under continuous stirring. Unstable albumin aggregates are formed and are hardened by chemical crosslinking to prevent redissolving.
|
Human serum nanoparticles containing antisense oligonucleotides [97]
Folate-decorated paclitaxel-loaded bovine serum albumin nanoparticles [98] |
| Emulsion | 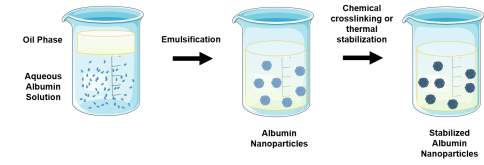 Dropwise addition of nonaqueous phase to aqueous albumin solution under continuous stirring. The resulting emulsion is then sonicated and a chemical crosslinking emulsion (e.g. glutaraldehyde) or high heat can be used to stabilize the resulting nanoparticles. Dropwise addition of nonaqueous phase to aqueous albumin solution under continuous stirring. The resulting emulsion is then sonicated and a chemical crosslinking emulsion (e.g. glutaraldehyde) or high heat can be used to stabilize the resulting nanoparticles. |
PEGylated albumin nanoparticles encapsulating azidothymidine [99]
10-hydroxycamptothecin-loaded bovine serum albumin nanoparticles [100] |
| Thermal Gelation | 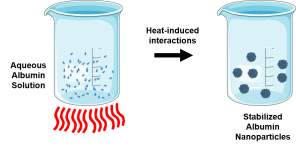 Heat-induced unfolding followed by protein-protein interactions such as hydrogen bonding, electrostatic, hydrophobic, and disulfide-sulfhydryl. Heat-induced unfolding followed by protein-protein interactions such as hydrogen bonding, electrostatic, hydrophobic, and disulfide-sulfhydryl. |
Bovine serum albumin-dextran-chitosan nanoparticles encapsulating doxorubicin [101] |
| Nanospraying | 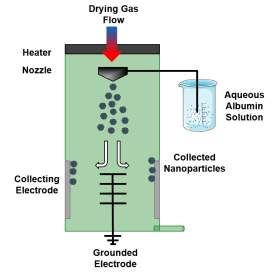 Drying gas enters system through heater. Nozzle sprays fine droplets with narrow size distribution into chamber where they dry in to solid particles. These particles are then collected using electrostatic particle collector consisting of grounded star electrode and cylindrical collecting electrode. Drying gas enters system through heater. Nozzle sprays fine droplets with narrow size distribution into chamber where they dry in to solid particles. These particles are then collected using electrostatic particle collector consisting of grounded star electrode and cylindrical collecting electrode. |
Bovine serum albumin nanoparticles [102] |
| Self-Assembly | 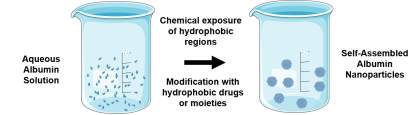 Increase hydrophobicity of albumin by breaking disulfide bonds, removing primary amines , or addition of lipophilic drugs. Increase hydrophobicity of albumin by breaking disulfide bonds, removing primary amines , or addition of lipophilic drugs. |
Inhalable human serum albumin nanoparticles conjugated with doxorubicin and adsorbed with TRAIL [103]
Mannosylated albumin nanoparticles [104] |
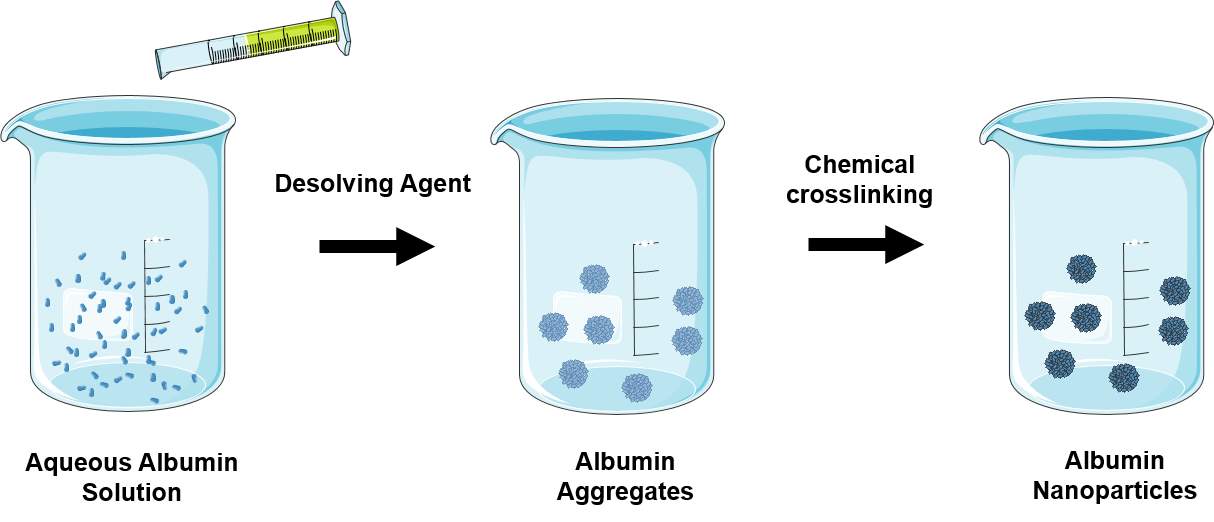
Albumin nanoparticles can be decorated with a variety of targeting ligands to give additional specificity to cancer-associated receptors. For instance, Zhao et al sought to target drug resistant colon-cancer cells and tumor associated macrophages which both highly express mannose receptors and SPARC [105]. They used mannosylated bovine serum albumin which was synthesized using a molar 4-isothiocyanatophenyla-α-mannopyranoside. The albumin was then denatured using urea/BH4 and the addition of hydrophobic drugs and changing salt concentration resulted in self-assembly into drug loaded nanoparticles. In an analogous approach, folate-decorated bovine serum albumin nanoparticles were developed for the targeted delivery of paclitaxel. This approach leverages the overexpression of folate receptor which is known to occur on a wide range of tumor cell types; importantly the folate receptor is continuously internalized as a mechanism for carrying folate (vitamin B9) into tumor cells and thus serves as a particular useful portal for cellular entry [106]. This receptor targeted nanoparticle was formulated by a desolvation method where BSA was dissolved into water followed by the addition of paclitaxel in ethanol using a peristaltic pump to drive aggregation. Then, a gluataraldehyde solution was added to crosslink the amino groups of the nanoparticles. After formation of the nanoparticles, an N-hydroxysuccinimide ester of folate was conjugated to free amines of BSA under alkaline conditions [98]. The resultant particles were approximately 210 nm in diameter and achieved a drug loading efficiency of approximately 27%. In addition to decorating albumin-based nanoparticles with natural receptor ligands, similar nanoparticle formulations have been targeted through functionalization with antibodies. One such example employed covalent coupling of DI17E6, a monoclonal antibody directed against αv integrins, which are cell membrane-spanning matrix adhesion domains that are highly expressed in a various cancer lines. Inhibitors of αvβ3 have been shown to inhibit growth and angiogenesis in melanoma. DI17E6-functionlized particles were formulated by ethanol desolvation of an aqueous solution of doxorubicin adsorbed to HSA followed by crosslinking with glutaraldehyde. Free amines on the surface of these particles were modified with thiolated antibody through a PEG maleimide-NHS ester. The resultant loaded drug particles showed very low dispersity at a size of approximately 380nm [107]. These examples underscore the versatility of albumin-based carriers for small molecule chemotherapeutics and also for modification to target specific cancer cell types.
Albumin nanoparticle composites have also shown efficacy in the encapsulation and delivery of anticancer agents. For example, silk fibroin-albumin blended nanoparticles have been developed with the goal of circumventing the leakage of encapsulated chemotherapeutics believed to occur due to the relatively hydrophilic nature of albumin [108]. It was hypothesized that because silk fibroin is a more hydrophobic protein, a composite nanoparticle would improve mechanical and encapsulation properties due to the electrostatic interactions between carboxyl groups of the silk fibroin and the amino groups of albumin. The authors applied this system to encapsulate the chemotherapeutic methotrexate into ~100 nm particles; this was achieved by desolvation in acetone and glutaraldehyde crosslinking of a silk fibroin and albumin solution [108]. Additionally, albumin has been derivatized into a diblock format with tranditionally-utilized, hydrolytically-degradable, and hydrophobic synthetic polymers such as polycaprolactone. In this setting, a micellar nanoparticle can be formed with the albumin serving as the hydrophilic block which is traditionally PEG. Jiang et al created two different polymers based on ring-opening polymerization: poly(oligo(ethylene glycol) methyl ether acrylate)–poly(ε-caprolactone) (POEGMA-PCL) and maleimide –functionalized polycaprolactone (MI-PCL). This maleimide PCL was then conjugated to the free cysteine on bovine serum albumin. The authors then synthesized nanoparticles with different albumin content through the co-assembly of POEGMA-PCL and BSA-PCL at different mixture ratios. They then loaded these particles with curcumin, and noted that for several cancer cell lines, cellular uptake of the nanoparticles positively correlated with the amount of albumin present on the nanoparticle surface [109]. Albumin modified nanoparticles may benefit from reduced of toxicity of their degradation products, increased ease of injectability, and solubility in water. This overall body of literature suggests that in conjunction with the previously discussed tumor-tropic properties of albumin, it is an ideal candidate for nanoparticle synthesis.
3. Survey of Results
Albumin has demonstrated benefit as a drug carrier in both preclinical models and clinical trials. In the following sections, we will highlight the ability of diverse strategies employing albumin to improve the pharmacokinetic properties of anticancer agents including biodistribution, circulation half-life, tumor targeting and penetration, and ultimately, tumor cytotoxicity and survival outcomes. Applications with in vivo results are preferentially highlighted in this section.
3.1 Chemotherapeutics
3.1.1 Nanoparticles
Albumin nanoparticles have been investigated in many cancer types, but are especially well-suited for the challenging cancers of the brain. Thebrain-blood barrier typically serves the important physiological purpose of protecting the brain from insults in the circulation. However, it also presents the greatest challenge for drug delivery to cancers of the brain. Despite the poor transport across the brain vascular endothelium, there is exchange of substances between the intravascular and tissue compartment of the brain through active nutrient transporters [110]. Lin and colleagues were motivated to leverage the increased demand for albumin from metabolically actively tumors to facilitate the intake of a drug-loaded albumin nanoparticle. Their self-assembled formulation included a cell penetrating peptide, low molecular weight protamine, on the outside of the particle to aid in tissue penetration and two hydrophobic chemotherapeutics, paclitaxel and fenretinide. Their nanoparticle demonstrated an inhibition rate of 82% in a subcutaneous glioma mouse model compared to 40% efficacy for the free combination of drugs. Furthermore, in an orthoptopic glioma (Luc-U87) model, their particle administered with 2 mg/kg of each of their therapeutics demonstrated a greater survival time of 37 days relative to 24 days for negative control and 31days for equivalent concentrations of free drug (Figure 5). Interestingly, protumor phenotype M2 macrophages were suppressed by their nanoparticles.
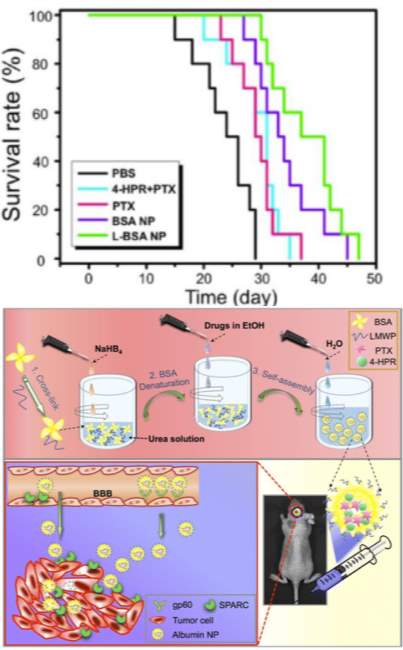
Figure 5: Top) Survival curve of mice bearing orthotopic gliomas treated with cell penetrating peptide modified (L-BSA-NP), unomodified nanoparticle (BSA-NP), free fenretinide (4-HPR), free paclitaxel (PTX), free drug combination (4-HPR+PTX), or phosphate buffered saline control (PBS). Bottom) Self-assembly of low molecular weight protamine-modified albumin nanoparticles. Figure reproduced with kind permission from Lin et al [111]
For direct pulmonary treatment of lung cancer, Choi and colleagues synthesized inhalable albumin nanoparticles made of human serum albumin conjugated with doxorubicin and octyl aldehyde and absorbed with TRAIL [103]. Briefly, this involved thiolating doxorubicin and modifying HSA with a maleimide (sulfo-SMCC linker). These two components were then reacted, and the resulting conjugate was functionalized with octanal by reductive amination in the presence of sodium cyanoborohydride followed by loading with TRAIL. These particles achieved anti-tumor efficacy in mice bearing lung H226 tumors, with lungs of mice treated with the TRAIL/Dox HSA nanoparticle (5µg TRAIL + 1µg Dox) showing fewer malignant surface lesions compared to mice treated with dose matched Dox or TRAIL HSA. Lung weight as a measurement of tumor expansion from groups treated with TRAIL particles or Dox particles was significantly greater than that of the combination TRAIL/Dox HSA-NP (678.2 ± 51.5, 598 .9 ± 24.8, and 337.5 ± 7.5 mg respectively) (Figure 6). Additionally, histological samples of the lung specimens from H226-implanted mice demonstrated that TRAIL/Dox HSA-NP not only decreased lesion numbers and sizes, but exhibited significant induction of apoptosis whereas mice treated with TRAIL or Dox particles did not. With lung cancer presently associated with a particularly low 5-year survival rate of less than 18%, high efficiency delivery of therapeutics directly to lung tumors using albumin presents a promising approach.
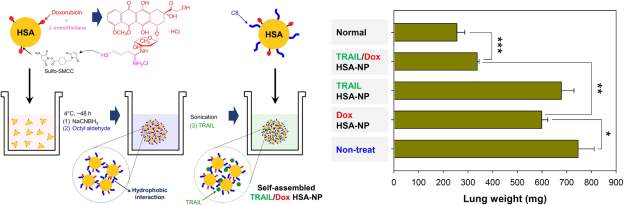
Figure 6: Synthesis overview of TRAIL/Dox HSA-NP and lung weight as a measure of tumor burden in treated mice. Figure reproduced with kind permission from Choi et al [103]
3.1.2 Covalent Conjugates
Chemotherapeutics directly bound to exogenous and endogenous albumin rather than encapsulated into nanoparticles have also demonstrated notable benefits. For the albumin-binding doxorubicin prodrug synthesized by Chung et al, the addition of an albumin-binding moiety resulted in the extension of half-life 38-fold from approximately 30 minutes to 19 hours. Doxorubicin was linked via a self-immolative linker to a DEVD motif. This DEVD-S-DOX conjugate was linked to the albumin-binding maleimide functionalization, ε-maleimidocaproic acid (EMC) using an N-succinimide ester. The DEVD spacer is recognized and hydrolyzed by caspase-3, a protease upregulated in irradiated tumors. The administration of this conjugate labeled with Cy5 in lieu of doxorubicin resulted in significant accumulation in tumors with fluorescent intensity decreasing in other organs. Notably, in vitro there was a lack of toxicity associated with HSA-DEVD-S-DOX up to 100µM until incubation with caspase 3, whereas, under these activated conditions, the prodrug created similar toxicity as free doxorubicin (IC50=0.49 and 4.17 µm and IC50=0.30 and 2.04 µM respectively). Furthermore, insignificant accumulation of the compound within the cell nucleus was observed without the presence of caspase-3, further underscoring the ability of the system to avoid off target effects using environmentally targeted activation. When administered at 10 mg/kg with radiotherapy, the authors reported a 91% decrease in tumor volume compared to radiotherapy alone with no noticeable body weight changes or indications of systemic toxicity. According to the results, EMC-DEVD-S-DOX had negligible anti-cancer effect when used alone, underscoring the ability of this prodrug to overcome the nonspecific toxicity shown in traditional chemotherapy [47].
The same benefits conferred by binding to endogenous albumin can be achieved by the covalent conjugation of exogenous albumin to chemotherapeutics. Notably, methotrexate conjugated at a 1:1 drug:HSA ratio through HSA lysine residues found significant success in preclinical animal models (clinical translatability reviewed in Section 3.4). Motivated by the short tumor exposure time of unmodified methotrexate, an albumin-MTX conjugate was developed to improve the MTX pharmacokinetic profile. These conjugates were evaluated in seven nude mouse human tumor xenograft models including bladder, breast, lung, osteosarcoma, soft tissue sarcoma, and prostate cancers [112]. Notably, in soft tissue sarcoma SXF 1301, MTX-HSA treatment resulted in complete remission after a single injection at 12.5 mg/kg whereas an equivalent drug dose of free MTX resulted in short-lasting, partial tumor regression. Additionally, in the prostate-cancer model PRXF PC3M, MTX-HSA demonstrated 92.8% growth inhibition of control. However, at a molar basis, the authors noted that MTX-HSA was more active than MTX but also more toxic. The authors cited the potential for this compound to overcome drug resistance due to transport deficiencies for native MTX, and suggested the improved pharmacokinetic properties, persistent high plasma levels, and demonstrated accumulation in solid tumors contributed to the potential efficacy of their compound.
3.2 Biologics
3.2.1 Oligonucleotides
Albumin-mediated delivery of therapeutic oligonucleotides has found success in multiple preclinical models of cancer. Conjugation of therapeutic short interfering siRNA via a PEGylated linker to an albumin-binding diacyl lipid (L2) prolonged the half-life of siRNA by 6-fold. Notably, siRNA molecules can be degraded by nucleases and are rapidly cleared through the kidneys following intravenous delivery, and association with albumin appears to reduce both of these clearance mechanisms. In addition to systemic pharmacokinetic advantages, testing on in vitro MCF7 breast cancer spheroids showed that albumin-bound siRNA conjugates have increased penetration and both higher and more homogenous tumor cell internalization than commercially available jetPEI nano-polyplexes (Figure 7). The siRNA-L2 conjugates also localized to tumors significantly more than commercially available jetPEI in a patient derived xenograft model at 1mg/kg (99% uptake vs. 60% uptake 30 minutes after i.v. injection), and tumor radiance as an indicator of Luciferase knockdown was significantly lower for this formulation than jetPEI in an orthotopic tumor of MDA-MB-231 cells. One striking finding from this study was a complete lack of toxicity at doses of 10mg/kg, which sharply contrasts with reported toxicity and immunogenicity of nanoparticle carriers. [55] Additionally, siRNA-L2 demonstrated a tumor to liver ratio of 40:1 compared to 3:1 for jetPEI. This finding is particularly notable given that a main disadvantage of nanoparticles is their natural tendency to accumulate in the liver. These data suggest that albumin-based nucleotide delivery may be especially promising for nonhepatic, oncologic therapeutic applications relative to synthetic nanocarriers.
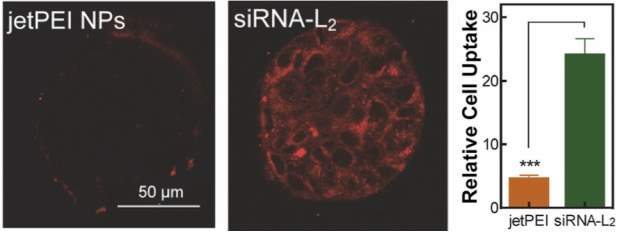
Figure 7: Diacyl modified siRNA (siRNA-L2) demonstrated superior tumor penetration and relative cell uptake to commercially available jetPEI. Figure reproduced with kind permission from Sarett et al [55]
Short interfering RNA has also been delivered to tumors via self-crosslinked human serum albumin nanocarriers (Figure 8) [113]. These nanocomplexes were comprised of thiolated HSA that would interact with thiolated siRNA to self-crosslink. Tail vein injection of 50 mg of Cy5-labeled nanocomplex to SCC7 tumor-bearing mice resulted in 1.7 times the signal intensity in tumor tissue 12 hours post-injection than thiolated siRNA alone. Using RFP/B16F10 tumor-bearing mice, Son and colleagues were able to show their nanoparticle/siRNA formulation turning off the RFP signals at the tumor site (RFP signal diminished to 43% at 7 days compared to saline control and free thiolated siRNA. The authors also quantified gene expression by RT-PCR and showed that the amount of RFP mRNA after treatment with their nanoparticle was decreased to about 19% of the control while free precursor thiolated siRNA showed no significant reduction. Finally, they demonstrated anti-tumor therapeutic efficacy of their nanoparticle formulation containing siRNA against vascular endothelial growth factor, which has been implicated as a key angiogenic factor that drives tumor angiogenesis and growth. Their treatment (0.5 mg of siRNA/kg once per 3 days by i.v. injection) resulted in the reduction of tumor volumes by 80% compared to the control group at day 30 (no significant reduction from free thiolated siRNA siRNA).
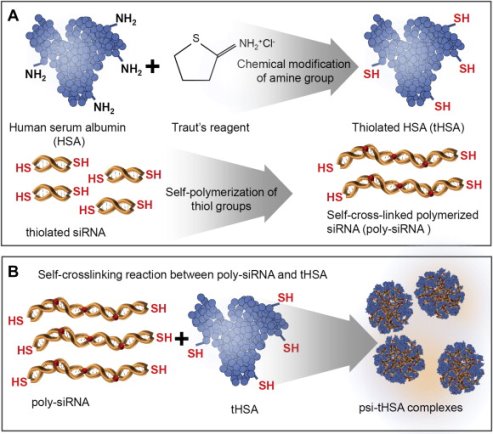
Figure 8: Thiol modified siRNA and albumin molecules are able to self-assemble into nanocomplexes by the formation of disulfide bridges. Figure reproduced with kind permission from Son et al [113]
Albumin-mediated delivery of antisense oligonucleotides (ASO) has also been investigated in the context of cancer treatment. For instance, Wartlick and colleagues first demonstrated uptake of ASO bearing albumin nanoparticles in the breast cancer cell lines MCF-7 and MDA-BM-435 administered at 1mg/mL [97]. Subsequent efforts to mediate ASO delivery with albumin have included the investigation of a folate receptor-targeted lipid-albumin nanoparticle for the delivery of Akt1, an ASO which has shown clinical efficacy in tumor inhibition but is limited by low membrane permeability and rapid clearance in the blood [114].
3.2.2 Immunomodulatory Drugs
Albumin mediated-delivery has also demonstrated success in the context of tumor immunotherapy. The albuleukin fusion peptide of recombinant interleukin-2 and human serum albumin showed the ability of genetic fusion to albumin to confer its remarkable pharmacokinetic properties [94]. Indeed, pharmacokinetic studies of 500µg/kg of albuleukin versus the rIL-2 peptide alone in BALC/c mice demonstrated half-life extension from 19-57 minutes to 6 to 8 hours. Importantly, the immunomodulatory effects associated with the rIL-2 peptide alone were maintained in the fusion peptide. In Renca renal adenocarninoma tumors in mice, albuleukin suppressed tumor growth significantly compared to rIL-2 alone and promoted infiltration of CD4+ and CD8+ T cells, which are associated with a beneficial anti-tumor immune response. Albuleukin additionally inhibited metastasis of hepatic tumors of B16F10 melanoma cells when administered to mice every other day at 1 mg/kg for three days with significant reduction in residual metastases compared to rIL-2 alone (~30 vs ~80 percent mice with residual metastases). Hepatic metastasis of melanoma continues to present a significant clinical challenge, underscoring the need for new treatment strategies such as albuleukin.
Despite the tremendous potential of vaccines in cancer immunotherapy, inefficient delivery of antigen and adjuvants to the lymph node, where the immune response of lymphocytes is coordinated, has limited their clinical success. At 67 kDa, albumin exceeds the cutoff for dissemination in to the blood from the interstitial space and is instead trafficked to the lymphatics [115]. Zhu et al sought to leverage this characteristic along with slow lymph flow to modulate lymphocytes within lymph nodes. Albumin can be efficiently endocytosed by antigen-presenting cells (APCs) via endocytosis which can facilitate intracellular vaccine delivery for optimal antigen processing and presentation. By conjugating molecular vaccines to Evans Blue, albumin-binding vaccines that self-assemble in vivo using endogenous albumin were found to be 100-fold more efficient at the co-delivery of CpG and antigens to lymph nodes. This albumin vaccine nanocomplex was additionally shown to elicit ~10 times more antigen-specific CD8+ cytotoxic T lymphocytes with immune memory than the benchmark incomplete Freund’s adjuvant. This formulation in conjunction with Abraxane or anti-PD 1 shows further potency and represents a robust platform for combination cancer immunotherapy [66].
3.3 Theranostics
The unique characteristics of albumin as a carrier lend themselves to utilization of this protein for theranostics, which can not only be used for diagnosis and imaging but also for treatment. For instance, lymph node metastasis is associated with poor prognosis. Chen and colleagues demonstrated they could create nanoprobes for image-guided photoablation by adsorbing IR825, a near-infrared dye, to human serum albumin covalently linked with diethylenetriamine pentaacetic acid molecules chelating gadolinium gadolinium. (Figure 9). Specifically, they demonstrated inhibition of tumor metastasis after surgery by photothermally ablating sentinel lymph nodes (SNLs) with metastatic tumor cells. In an in vitro study, 4T1 murine breast cancer cells efficiently internalized HSA-Gd-IR825 nanoprobes with no difference in cell viability until photothermal ablation was induced. Mice inoculated with 4T1 murine breast tumor cells via footpad injection developed metastatic tumor cells in their sentinel lymph nodes after 12 days. When 0.03µmol of HSA-Gd-IR825 was administered intratumorally on the right foot pad, these sentinel lymph nodes were rapidly heated upon exposure to an 808 nm laser at a power density of 0.8 W/cm2 for 10 min. For mice injected with the nanoprobe, temperatures of the SLNs reached ~55°C: high enough to ensure effective photothermal ablation of cancer cells, whereas saline injected mice under the same irradiation conditions showed little change. Excitingly, the researchers found that mice that underwent surgical dissection of their primary tumors as well as photothermal ablation of their SNLs showed greatly prolonged survival. Notably, 4 out of 6 mice remained alive after 70 days compared to mice with primary tumor isolation alone dying within 50 days. Such a strategy could be an alternative approach to assist surgery and reduce the risk of post-treatment metastasis via lymphatic systems [116].
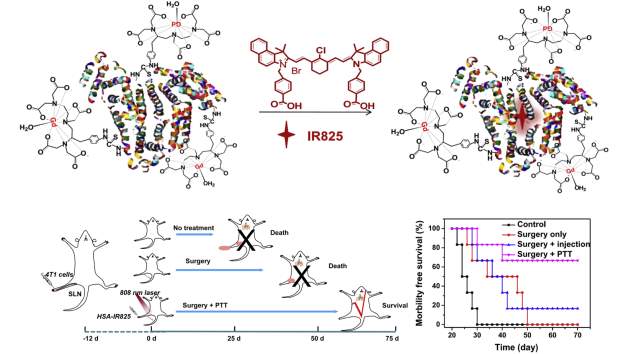
Figure 9: Albumin-mediated photothermal ablation (PTT) of metastasized areas results in prolonged morbility free survival time in 4T1 mouse tumor model. Figure reproduced with kind permission from Chen et al [116]
Another exciting approach from Chen et al utilized a nanoscale delivery system based on albumin-coated MnO2, which can modulate the tumor microenvironment by relieving hypoxia [117]. It is known that the rapid growth of cancer cells results in insufficient blood supply to tumors, and the resultant hypoxia may hinder treatments such as oxygen-dependent radiotherapy and photodynamic therapy. Manganese dioxide (MnO2) nanoparticles are known to have high reactivity toward H2O2 (solid tumors constitutively produce H2O2) to produce O2. By leveraging MnO2 decomposition in the reactive oxygen species rich tumor condition of the tumor, the authors sought to overcome tumor-hypoxia associated resistance of photodynamic therapy. Importantly, the tumor growth in 4T1 tumor bearing nude mice after combination therapy using the HSA MnO2 nanoparticles plus 660 nm light irradiation was significantly inhibited. The HSA MnO2 system provided improved therapeutic efficacy compared to various control groups, including the combination therapy using HSA NPs plus light irradiation without the assistance of MnO2 to relieve tumor hypoxia.
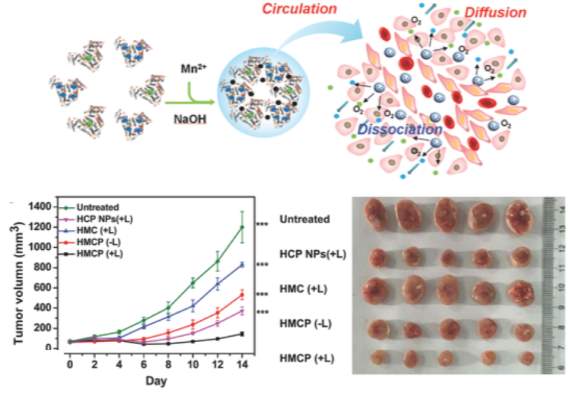
Figure 10
Top:Overview of MnO2 albumin nanoparticle mechanism. Bottom: Tumor growth curves of mice after various treatments (multicomponent HSA‐MnO2‐Ce6&Pt (HMCP)
light irradiation indicated by L+). Photographs of tumors collected from different groups of mice 14 d after treatment. Figure reproduced with kind permission from Chen et al [117]
3.4 Clinical Results
The promising results from preclinical investigation of albumin-mediated cancer treatments have prompted subsequent clinical trials. The first albumin-drug conjugate evaluated in phase I/II studies was the aforementioned methotrexate-albumin conjugate synthesized by the direct covalent conjugation to lysine residues of HSA. Seventeen patients were treated, and tumor responses were seen in three: partial and minor responses in patients with renal cell carcinoma and pleural cell carcinoma. A regimen with MTX-HSA injections of 50 mg/m2 every 2 weeks was recommended for further investigation after this study [118]. However, a subsequent phase II study did not show any objective responses despite being well tolerated (n=17) [119]. A phase II study assessing administration in combination with cisplatin showed one complete response and one partial response in advanced bladder cancer (n=7) but no further clinical investigation was undertaken [120].
Phase 1 clinical studies of the (6-maleimidocaproyl) hydrazone derivative of doxorubicin (aldoxorubicin) showed a good safety profile at doses up to 260 mg/m2 and induced tumor regression in breast cancer, small cell lung cancer, and sarcoma [121]. In 2015, aldoxorubicin was investigated in a phase 2b clinical trial for advanced soft-tissue sarcoma. A total of 123 patients were treated with 83 receiving aldoxorubicin at 350 mg/m2 or doxorubicin at 75 mg/m2 once every three weeks for up to 6 cycles. Median progression-free survival time was significantly improved (5.6 vs 2.7 months) with aldoxorubicin compared to doxorubicin as well as the rate of 6-month progression-free survival (46% and 23%) [122]. The investigators concluded that further investigation of aldoxorubicin therapy in advanced soft-tissue sarcoma is warranted.
Abraxane (nab-paclitaxel) was the first albumin-based drug approved in oncology. It was first approved in 2005 for the treatment of metastatic breast cancer and subsequently approved in 2012 and 2013 for first-line treatment of non-small cell lung cancer and metastatic pancreatic cancer, respectively [123]. The design of the phase III studies for both of was similar: Abraxane was combined with the best chemotherapeutic agent of choice and this combination was compared with the single standard agent alone (100mg/m2 with carboplatin every 3 weeks for non-small cell lung cancer compared to cremophor encapsulated paclitaxel plus carboplatin once every three weeks). For advanced pancreatic cancer, 125 mg/m2 Abraxane was combined with 1000mg/m2 gemcitabine compared with the gemcitabine monotherapy on a three-week schedule. The outcome of these studies showed ~1 month (NSCLC) and ~2 month (pancreatic cancer) benefits for the combination therapy.
3.5 Conclusions and Future Directions
In sum, albumin-mediated delivery has demonstrated advantages for multiple categories of anticancer agents. Its cancer targeting properties, extraordinary half-life, and natural ligand binding capability make it a promising and already translatable medium. Excitingly, many high affinity albumin-binding platforms described in the synthesis portion of this review have yet to be examined in the context of cancer treatment. Moreover, as our understanding of molecular targets for treating the diverse spectrum of cancer variations develops, one can imagine the more tailored and personalized medicine that may be made possible through albumin-mediated delivery of biologics and immunomodulatory drugs.
Moving forward, it will be necessary to characterize the cancer subsets that are most amenable to albumin-mediated treatment by potentially examining expression of Cav-1 as well as by characterizing vasculature. Further investigation is also warranted to provide a better understanding of the fate of drugs bound to albumin after transcytosis and target uptake. The diverse array of albumin binders reviewed may affect the mechanisms of delivery, and a comparative study of different albumin-based formulations is highly warranted. Albumin is a remarkable carrier protein with the potential to overcome barriers to delivering a host of previously hindered but promising therapeutics in the cancer arena thanks to its half-life extending properties, preferential tumor accumulation and penetration, and natural ligand transporting ability.
References
[1] M. T. Larsen, M. Kuhlmann, M. L. Hvam, and K. A. Howard, “Albumin-based drug delivery: harnessing nature to cure disease,” Mol. Cell. Ther., vol. 4, no. 1, p. 3, Dec. 2016.
[2] “Albumin as a versatile platform for drug half-life extension,” Biochim. Biophys. Acta – Gen. Subj., vol. 1830, no. 12, pp. 5526–5534, Dec. 2013.
[3] D. Carter and J. Ho, “Structure of Serum Albumin,” Adv. Protein Chem., vol. 45, 1994.
[4] U. Kragh-Hansen, “Molecular aspects of ligand binding to serum albumin.,” Pharmacol. Rev., vol. 33, no. 1, 1981.
[5] E. Neumann and C. Fiehn, “Native albumin for targeted drug delivery,” Expert Drug Deliv., 2010.
[6] V. Arroyo, R. García-Martinez, and X. Salvatella, “Human serum albumin, systemic inflammation, and cirrhosis,” J. Hepatol., vol. 61, no. 2, pp. 396–407, Aug. 2014.
[7] E. Schnitzers and P. Oh, “Albondin-mediated Capillary Permeability to Albumin DIFFERENTIAL ROLE OF RECEPTORS IN ENDOTHELIAL TRANSCYTOSIS AND ENDOCYTOSIS OF NATIVE AND MODIFIED ALBUMINS*,” J. Biol. Chem., vol. 269, no. 8, pp. 6072–6082, 1994.
[8] A. M. Merlot, D. S. Kalinowski, D. R. Richardson, and B. Vogt, “MINI REVIEW ARTICLE Unraveling the mysteries of serum albumin—more than just a serum protein,” 2014.
[9] H. Birn et al., “Cubilin is an albumin binding protein important for renal tubular albumin reabsorption.,” J. Clin. Invest., vol. 105, no. 10, pp. 1353–61, May 2000.
[10] H. Birn and E. I. Christensen, “Renal albumin absorption in physiology and pathology,” Kidney Int., vol. 69, no. 3, pp. 440–449, Feb. 2006.
[11] S. Amsellem et al., “Cubilin is essential for albumin reabsorption in the renal proximal tubule.,” J. Am. Soc. Nephrol., vol. 21, no. 11, pp. 1859–67, Nov. 2010.
[12] N. J. Ganson et al., “Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer.,” J. Allergy Clin. Immunol., vol. 137, no. 5, p. 1610–1613.e7, May 2016.
[13] R. K. Jain and T. Stylianopoulos, “Delivering nanomedicine to solid tumors,” Nat. Rev. Clin. Oncol., vol. 7, no. 11, pp. 653–664, Nov. 2010.
[14] N. Bertrand, J. Wu, X. Xu, N. Kamaly, and O. Farokhzad, “Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology,” Adv. Drug Deliv. Rev., vol. 66, 2013.
[15] Y. Matsumura and H. Maeda, “A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs.,” Cancer Res., vol. 46, no. 12 Pt 1, pp. 6387–92, Dec. 1986.
[16] T. W. Evans, “Review article: albumin as a drug-biological effects of albumin unrelated to oncotic pressure,” Aliment. Pharmacol. Ther., vol. 16, no. s5, pp. 6–11, Dec. 2002.
[17] L. Tang et al., “Investigating the optimal size of anticancer nanomedicine.”
[18] G. Stehle et al., “Hematology Plasma protein (albumin) catabolism by the tumor itself-implications for tumor metabolism and the genesis of cachexia,” Crit. Rev. Oncol. Hematol., vol. 26, pp. 77–100, 1997.
[19] K. M. Knudsen Sand, M. Bern, J. Nilsen, H. T. Noordzij, I. Sandlie, and J. T. Andersen, “Unraveling the interaction between FcRn and albumin: Opportunities for design of albumin-based therapeutics,” Frontiers in Immunology, vol. 6, no. JAN. 2015.
[20] M. A. Miller et al., “Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle.,” Sci. Transl. Med., vol. 7, no. 314, p. 314ra183, Nov. 2015.
[21] S. Qin et al., “A physiological perspective on the use of imaging to assess the in vivo delivery of therapeutics,” Ann. Biomed. Eng., vol. 42, no. 2, pp. 280–298, 2014.
[22] P. M. Campbell and C. J. Der, “Oncogenic Ras and its role in tumor cell invasion and metastasis,” Semin. Cancer Biol., vol. 14, no. 2, pp. 105–114, Apr. 2004.
[23] C. Commisso et al., “Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells,” Nature, vol. 497, no. 7451, pp. 633–637, May 2013.
[24] J. J. Kamphorst et al., “Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein,” Cancer Res., vol. 75, no. 3, pp. 544–553, 2015.
[25] C. A. Hauser, M. R. Stockler, and M. H. N. Tattersall, “Prognostic factors in patients with recently diagnosed incurable cancer: a systematic review,” Support Care Cancer, vol. 14, pp. 999–1011, 2006.
[26] G. Stehle et al., “Plasma protein (albumin) catabolism by the tumor itself—implications for tumor metabolism and the genesis of cachexia,” Crit. Rev. Oncol. Hematol., vol. 26, no. 2, pp. 77–100, Jul. 1997.
[27] M. Chatterjee et al., “Caveolae-Mediated Endocytosis Is Critical for Albumin Cellular Uptake and Response to Albumin-Bound Chemotherapy.”
[28] N. Desai, V. Trieu, B. Damascelli, and P. Soon-Shiong, “SPARC Expression Correlates with Tumor Response to Albumin-Bound Paclitaxel in Head and Neck Cancer Patients,” Transl. Oncol., vol. 2, no. 2, pp. 59–64, Jun. 2009.
[29] M. Hidalgo et al., “Hidalgo MPACT SPARC Results; Final v1 SPARC Expression Did Not Predict Efficacy of nab-Paclitaxel Plus Gemcitabine or Gemcitabine Alone for Metastatic Pancreatic Cancer in an Exploratory Analysis of the Phase III MPACT Trial.”
[30] T. M. Williams and M. P. Lisanti, “Caveolin-1 in oncogenic transformation, cancer, and metastasis,” Am. J. Physiol. Physiol., vol. 288, no. 3, pp. C494–C506, Mar. 2005.
[31] B. Razani, S. E. Woodman, and M. P. Lisanti, “Caveolae: From Cell Biology to Animal Physiology.”
[32] M. Gumbleton, “Caveolae as potential macromolecule trafficking compartments within alveolar epithelium.,” Adv. Drug Deliv. Rev., vol. 49, no. 3, pp. 281–300, Jul. 2001.
[33] S. M. R. Pinilla, E. Honrado, D. Hardisson, J. Benítez, and J. Palacios, “Caveolin-1 expression is associated with a basal-like phenotype in sporadic and hereditary breast cancer,” Breast Cancer Res. Treat., vol. 99, no. 1, pp. 85–90, Sep. 2006.
[34] S. A. Tahir et al., “Secreted caveolin-1 stimulates cell survival/clonal growth and contributes to metastasis in androgen-insensitive prostate cancer.,” Cancer Res., vol. 61, no. 10, pp. 3882–5, May 2001.
[35] M. Suzuoki et al., “Impact of caveolin-1 expression on prognosis of pancreatic ductal adenocarcinoma,” Br. J. Cancer, vol. 87, no. 10, pp. 1140–1144, Nov. 2002.
[36] W. Schubert, P. G. Frank, B. Razani, D. S. Park, C. W. Chow, and M. P. Lisanti, “Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo.,” J. Biol. Chem., vol. 276, no. 52, pp. 48619–22, Dec. 2001.
[37] F. Kratz, R. Mü Ller-Driver, I. Hofmann, J. Drevs, and C. Unger, “A Novel Macromolecular Prodrug Concept Exploiting Endogenous Serum Albumin as a Drug Carrier for Cancer Chemotherapy.”
[38] † André Warnecke, ‡ Iduna Fichtner, # Dirk Garmann, # and Ulrich Jaehde, and † Felix Kratz*, “Synthesis and Biological Activity of Water-Soluble Maleimide Derivatives of the Anticancer Drug Carboplatin Designed as Albumin-Binding Prodrugs,” 2004.
[39] A. W. and and F. Kratz*, “Maleimide-oligo(ethylene glycol) Derivatives of Camptothecin as Albumin-Binding Prodrugs: Synthesis and Antitumor Efficacy,” 2003.
[40] V. Pichler et al., “Maleimide-functionalised platinum(iv) complexes as a synthetic platform for targeted drug delivery,” Chem. Commun., vol. 49, no. 22, p. 2249, Feb. 2013.
[41] F. Kratz et al., “Albumin Conjugates of the Anticancer Drug Chlorambucil: Synthesis, Characterization, andIn Vitro Efficacy,” Arch. Pharm. (Weinheim)., vol. 331, no. 2, pp. 47–53, Feb. 1998.
[42] C. Fiehn, F. Kratz, G. Sass, U. Müller-Ladner, and E. Neumann, “Targeted drug delivery by in vivo coupling to endogenous albumin: an albumin-binding prodrug of methotrexate (MTX) is better than MTX in the treatment of murine collagen-induced arthritis,” 2008.
[43] S. Lau, B. Graham, N. Cao, B. J. Boyd, C. W. Pouton, and P. J. White, “Enhanced Extravasation, Stability and in Vivo Cardiac Gene Silencing via in Situ siRNA−Albumin Conjugation.”
[44] H.-B. Pang et al., “A free cysteine prolongs the half-life of a homing peptide and improves its tumor-penetrating activity,” J. Control. Release, vol. 175, pp. 48–53, Feb. 2014.
[45] Y. Gou et al., “Developing Anticancer Copper(II) Pro-drugs Based on the Nature of Cancer Cells and the Human Serum Albumin Carrier IIA Subdomain,” Mol. Pharm., vol. 12, no. 10, pp. 3597–3609, Oct. 2015.
[46] Y. Zhang et al., “Structural basis and anticancer properties of ruthenium-based drug complexed with human serum albumin,” Eur. J. Med. Chem., vol. 86, pp. 449–455, Oct. 2014.
[47] S. W. Chung et al., “Albumin-binding caspase-cleavable prodrug that is selectively activated in radiation exposed local tumor,” Biomaterials, vol. 94, pp. 1–8, Jul. 2016.
[48] †,§ Björn Schmid, †,§ Da-Eun Chung, † André Warnecke, ‡ and Iduna Fichtner, and † Felix Kratz*, “Albumin-Binding Prodrugs of Camptothecin and Doxorubicin with an Ala-Leu-Ala-Leu-Linker That Are Cleaved by Cathepsin B: Synthesis and Antitumor Efficacy,” 2007.
[49] A. M. Mansour et al., “A new approach for the treatment of malignant melanoma: enhanced antitumor efficacy of an albumin-binding doxorubicin prodrug that is cleaved by matrix metalloproteinase 2.,” Cancer Res., vol. 63, no. 14, pp. 4062–6, Jul. 2003.
[50] J. Lau et al., “Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide,” J. Med. Chem., vol. 58, no. 18, pp. 7370–7380, Sep. 2015.
[51] H. Liu et al., “Structure-based programming of lymph-node targeting in molecular vaccines.,” Nature, vol. 507, no. 7493, pp. 519–22, Mar. 2014.
[52] M. L. Hvam et al., “Fatty Acid-Modified Gapmer Antisense Oligonucleotide and Serum Albumin Constructs for Pharmacokinetic Modulation,” 2017.
[53] A. Lacroix, T. G. W. Edwardson, M. A. Hancock, M. D. Dore, and H. F. Sleiman, “Development of DNA Nanostructures for High-Affinity Binding to Human Serum Albumin.”
[54] K. Madsen et al., “Structure-Activity and Protraction Relationship of Long-Acting Glucagon-like Peptide-1 Derivatives: Importance of Fatty Acid Length, Polarity, and Bulkiness,” 2007.
[55] S. M. Sarett et al., “Lipophilic siRNA targets albumin in situ and promotes bioavailability, tumor penetration, and carrier-free gene silencing.,” Proc. Natl. Acad. Sci. U. S. A., vol. 114, no. 32, pp. E6490–E6497, Aug. 2017.
[56] M. Van Woensel and S. De Vleeschouwer, “Development of siRNA-loaded chitosan nanoparticles targeting Galectin-1 for the treatment of glioblastoma mulitforme via intranasal adminstration,” J. Control. Release, 2016.
[57] K. Bienk et al., “An albumin-mediated cholesterol design-based strategy for tuning siRNA pharmacokinetics and gene silencing,” J. Control. Release, vol. 232, pp. 143–151, Jun. 2016.
[58] I. V. Chernikov et al., “Cholesterol-Containing Nuclease-Resistant siRNA Accumulates in Tumors in a Carrier-free Mode and Silences MDR1 Gene,” Mol. Ther. – Nucleic Acids, vol. 6, pp. 209–220, Mar. 2017.
[59] M. I. M. Natalya S. Petrova, Ivan V. Chernikov, IIya S. Dovydenko1, Aliya G. Venyaminova1, V. V. V. and Marina A. Zenkova, and Elena L. Chernolovskaya*, “Carrier-free cellular uptake and the gene-silencing activity of the lipophilic siRNAs is strongly affected by the length of the linker between siRNA and lipophilic group,” Nucleic Acids Res., vol. 40, no. 5, 2012.
[60] O. Jacobson, D. O. Kiesewetter, and X. Chen, “Albumin-Binding Evans Blue Derivatives for Diagnostic Imaging and Production of Long-Acting Therapeutics.”
[61] H. Chen et al., “Novel "Add-On" Molecule Based on Evans Blue Confers Superior Pharmacokinetics and Transforms Drugs to Theranostic Agents.,” J. Nucl. Med., vol. 58, no. 4, pp. 590–597, Apr. 2017.
[62] G. Niu et al., “In Vivo Labeling of Serum Albumin for PET.,” J. Nucl. Med., vol. 55, no. 7, pp. 1150–6, Jul. 2014.
[63] E. B. Ehlerding, X. Lan, and W. Cai, “"Albumin Hitchhiking" with an Evans Blue Analog for Cancer Theranostics.,” Theranostics, vol. 8, no. 3, pp. 812–814, 2018.
[64] H. Chen et al., “Chemical Conjugation of Evans Blue Derivative: A Strategy to Develop Long-Acting Therapeutics through Albumin Binding,” Theranostics, vol. 6, no. 62, 2016.
[65] Y. Liu et al., “Stable Evans Blue Derived Exendin‑4 Peptide for Type 2 Diabetes Treatment.”
[66] G. Zhu et al., “Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy.”
[67] C. E. Dumelin et al., “A Portable Albumin Binder from a DNA-Encoded Chemical Library,” Angew. Chemie Int. Ed., vol. 47, no. 17, pp. 3196–3201, Apr. 2008.
[68] S. Trüssel, C. Dumelin, K. Frey, A. Villa, F. Buller, and D. Neri, “New Strategy for the Extension of the Serum Half-Life of Antibody Fragments,” Bioconjug. Chem., vol. 20, no. 12, pp. 2286–2292, Dec. 2009.
[69] C. Müller, H. Struthers, C. Winiger, K. Zhernosekov, and R. Schibli, “DOTA conjugate with an albumin-binding entity enables the first folic acid-targeted 177Lu-radionuclide tumor therapy in mice.,” J. Nucl. Med., vol. 54, no. 1, pp. 124–31, Jan. 2013.
[70] R. B. Lauffer et al., “MS-325: Albumin-targeted Contrast Agent for MR Angiography,” Radiology, 1998.
[71] H. Ersoy and F. J. Rybicki, “Biochemical safety profiles of gadolinium-based extracellular contrast agents and nephrogenic systemic fibrosis,” J. Magn. Reson. Imaging, vol. 26, no. 5, pp. 1190–1197, 2007.
[72] T. Su et al., “Enhancing the circulating half-life and the antitumor effects of a tumor-selective cytotoxic peptide by exploiting endogenous serum albumin as a drug carrier,” Int. J. Pharm., vol. 499, no. 1–2, pp. 195–204, Feb. 2016.
[73] R. Li et al., “Fusion to an albumin-binding domain with a high affinity for albumin extends the circulatory half-life and enhances the in vivo antitumor effects of human TRAIL,” 2016.
[74] A. Jonsson, J. Dogan, N. Herne, L. Abrahmsen, and P.-A. Nygren, “Engineering of a femtomolar affinity binding protein to human serum albumin,” Protein Eng. Des. Sel., vol. 21, no. 8, pp. 515–527, May 2008.
[75] U. L. F. Sjobring, C. Falkenberg, E. Nielsen, B. O. Akerstrom, and L. Bjorck, “Isolation and characterization of a 14-kDa albumin-binding fragment of streptococcal Abbreviation used in this paper : HSA . human serum albumin .,” 2018.
[76] A. Binding et al., “Extended in Vivo Half-Life of Human Soluble Complement Receptor Type I Fused to a Serum Albumin-Binding Receptor,” J. Pharmacol. Exp. Ther., pp. 534–542, 1996.
[77] B. Hammarberg et al., “Dual affinity fusion approach and its use to express recombinant human insulin-like growth factor II.,” Proc. Natl. Acad. Sci. U. S. A., vol. 86, no. 12, pp. 4367–71, 1989.
[78] C. Libon et al., “The serum albumin-binding region of streptococcal protein G (BB) potentiates the immunogenicity of the G130-230 RSV-A protein,” Vaccine, vol. 17, no. 5, pp. 406–414, 1999.
[79] B. M. Tijink et al., “Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology.,” Mol. Cancer Ther., vol. 7, no. 8, pp. 2288–97, Aug. 2008.
[80] T. Yokota, D. E. Milenic, M. Whitlow, and J. Schlom, “Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms.,” Cancer Res., vol. 52, no. 12, pp. 3402–8, Jun. 1992.
[81] J. Löfblom, J. Feldwisch, V. Tolmachev, J. Carlsson, S. Ståhl, and F. Y. Frejd, “Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications,” FEBS Lett., vol. 584, no. 12, pp. 2670–2680, Jun. 2010.
[82] A. Orlova et al., “Site-specific radiometal labeling and improved biodistribution using ABY-027, a novel HER2-targeting affibody molecule-albumin-binding domain fusion protein.,” J. Nucl. Med., vol. 54, no. 6, pp. 961–8, Jun. 2013.
[83] M. Malm et al., “Engineering of a bispecific affibody molecule towards HER2 and HER3 by addition of an albumin-binding domain allows for affinity purification and in vivo half-life extension,” Biotechnol. J., vol. 9, no. 9, pp. 1215–1222, Sep. 2014.
[84] R. Stork, D. Muller, and R. E. Kontermann, “A novel tri-functional antibody fusion protein with improved pharmacokinetic properties generated by fusing a bispecific single-chain diabody with an albumin-binding domain from streptococcal protein G,” Protein Eng. Des. Sel., vol. 20, no. 11, pp. 569–576, Oct. 2007.
[85] A. M. Burger, G. Hartung, G. Stehle, H. Sinn, and H. H. Fiebig, “Pre-clinical evaluation of a methotrexate-albumin conjugate (MTX-HSA) in human tumor xenograftsin vivo,” Int. J. Cancer, vol. 92, no. 5, pp. 718–724, Jun. 2001.
[86] Biochemical, Biophysical, and Of, “Synthesis, Characterization and Anti-Cervical Cancer Cell Properties of Bovine Serum Albumen Curcumin Conjugate,” Biochem. Biophys. J. Neutron Ther. Cancer Treat., pp. 2331–8368, 2014.
[87] G. Di Stefano et al., “Efficacy of doxorubicin coupled to lactosaminated albumin on rat hepatocellular carcinomas evaluated by ultrasound imaging,” Dig. Liver Dis., vol. 40, no. 4, pp. 278–284, Apr. 2008.
[88] M. Kuhlmann and K. A. Howard, “An albumin-oligonucleotide assembly for potential combinatorial drug delivery and half-life estension applications,” Mol. Ther. Nucleic Acid, 2017.
[89] F. L. Pruitt, N. Brennen, L. Antony, D. M. Rosen, S. Denmeade, and J. Isaacs, “Abstract 2076: Albumin-linked proaerolysin based molecular grenades: A systemic therapeutic for disseminated castration resistant prostate cancer,” Cancer Res., vol. 76, no. 14 Supplement, pp. 2076–2076, Jul. 2016.
[90] R. J. Melder et al., “Pharmacokinetics and in vitro and in vivo anti-tumor response of an interleukin-2-human serum albumin fusion protein in mice.”
[91] C. Sung et al., “An IFN-b-Albumin Fusion Protein That Displays Improved Pharmacokinetic and Pharmacodynamic Properties in Nonhuman Primates,” J. Interf. CYTOKINE Res., vol. 23, pp. 25–36, 2003.
[92] P. J. Yazaki et al., “Biodistribution and tumor imaging of an anti-CEA single-chain antibody-albumin fusion protein.,” Nucl. Med. Biol., vol. 35, no. 2, pp. 151–8, Feb. 2008.
[93] J. Marques, J. George, I. Smith, V. Bhakta, and W. Sheffield, “A Barbourin-albumin Fusion Protein that is Slowly Cleared In Vivo Retains the Ability to Inhibit Platelet Aggregation In Vitro,” Thromb Haemost, vol. 86, pp. 902–8, 2001.
[94] A. O. Elzoghby, W. M. Samy, and N. A. Elgindy, “Albumin-based nanoparticles as potential controlled release drug delivery systems,” J. Control. Release, vol. 157, no. 2, pp. 168–182, Jan. 2012.
[95] B. Elsadek and F. Kratz, “Impact of albumin on drug delivery — New applications on the horizon,” J. Control. Release, vol. 157, no. 1, pp. 4–28, Jan. 2012.
[96] N. Desai et al., “Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel.,” Clin. Cancer Res., vol. 12, no. 4, pp. 1317–24, Feb. 2006.
[97] H. Wartlick, B. Spänkuch-Schmitt, K. Strebhardt, J. Kreuter, and K. Langer, “Tumour cell delivery of antisense oligonuclceotides by human serum albumin nanoparticles,” J. Control. Release, vol. 96, no. 3, pp. 483–495, May 2004.
[98] D. Zhao et al., “Preparation, characterization, and in vitro targeted delivery of folate-decorated paclitaxel-loaded bovine serum albumin nanoparticles.,” Int. J. Nanomedicine, vol. 5, pp. 669–77, Sep. 2010.
[99] V. Mishra et al., “Targeted brain delivery of AZT via transferrin anchored pegylated albumin nanoparticles.”
[100] L. Yang, F. Cui, D. Cun, A. Tao, K. Shi, and W. Lin, “Preparation, characterization and biodistribution of the lactone form of 10-hydroxycamptothecin (HCPT)-loaded bovine serum albumin (BSA) nanoparticles,” Int. J. Pharm., vol. 340, no. 1–2, pp. 163–172, Aug. 2007.
[101] J. Qi, P. Yao, F. He, C. Yu, and C. Huang, “Nanoparticles with dextran/chitosan shell and BSA/chitosan core—Doxorubicin loading and delivery,” Int. J. Pharm., vol. 393, no. 1–2, pp. 177–185, Jun. 2010.
[102] “Nano spray drying: A novel method for preparing protein nanoparticles for protein therapy,” Int. J. Pharm., vol. 403, no. 1–2, pp. 192–200, Jan. 2011.
[103] S. H. Choi et al., “Inhalable self-assembled albumin nanoparticles for treating drug-resistant lung cancer,” J. Control. Release, vol. 197, pp. 199–207, Jan. 2015.
[104] P. Zhao et al., “Dual-Targeting to Cancer Cells and M2 Macrophages via Biomimetic Delivery of Mannosylated Albumin Nanoparticles for Drug-Resistant Cancer Therapy,” Adv. Funct. Mater., vol. 27, no. 44, p. 1700403, Nov. 2017.
[105] P. Zhao et al., “Dual-Targeting to Cancer Cells and M2 Macrophages via Biomimetic Delivery of Mannosylated Albumin Nanoparticles for Drug-Resistant Cancer Therapy,” Adv. Funct. Mater., vol. 27, no. 44, p. 1700403, Nov. 2017.
[106] D. Zhao et al., “Preparation, characterization, and in vitro targeted delivery of folate-decorated paclitaxel-loaded bovine serum albumin nanoparticles.,” Int. J. Nanomedicine, vol. 5, pp. 669–77, Sep. 2010.
[107] S. Wagner et al., “Enhanced drug targeting by attachment of an anti αv integrin antibody to doxorubicin loaded human serum albumin nanoparticles,” Biomaterials, vol. 31, no. 8, pp. 2388–2398, Mar. 2010.
[108] B. Subia and S. C. Kundu, “Drug loading and release on tumor cells using silk fibroin–albumin nanoparticles as carriers,” Nanotechnology, vol. 24, no. 3, p. 35103, Jan. 2013.
[109] Y. Jiang, S. Wong, F. Chen, T. Chang, H. Lu, and M. H. Stenzel, “Influencing Selectivity to Cancer Cells with Mixed Nanoparticles Prepared from Albumin–Polymer Conjugates and Block Copolymers,” Bioconjug. Chem., vol. 28, no. 4, pp. 979–985, Apr. 2017.
[110] T. Lin et al., “Blood−Brain-Barrier-Penetrating Albumin Nanoparticles for Biomimetic Drug Delivery via Albumin-Binding Protein Pathways for Antiglioma Therapy.”
[111] T. Lin et al., “Blood–Brain-Barrier-Penetrating Albumin Nanoparticles for Biomimetic Drug Delivery via Albumin-Binding Protein Pathways for Antiglioma Therapy,” ACS Nano, vol. 10, no. 11, pp. 9999–10012, Nov. 2016.
[112] A. M. B. Urger, G. H. Artung, G. S. Tehle, S. Inn, and H. H. F. Iebig, “Pre-Clinical Evaluation of a Methotrexate – Albumin Conjugate ( Mtx-Hsa ) in Human Tumor Xenografts in Vivo,” vol. 724, no. March, pp. 718–724, 2001.
[113] S. Son et al., “Self-crosslinked human serum albumin nanocarriers for systemic delivery of polymerized siRNA to tumors,” Biomaterials, vol. 34, no. 37, pp. 9475–9485, 2013.
[114] H. Li et al., “Folate receptor-targeted lipid-albumin nanoparticles (F-LAN) for therapeutic delivery of an Akt1 antisense oligonucleotide,” J. Drug Target., vol. 0, no. 0, pp. 1–8, 2018.
[115] T. J. Moyer, A. C. Zmolek, and D. J. Irvine, “Beyond antigens and adjuvants: formulating future vaccines,” J. Clin. Invest., vol. 126, no. 3, pp. 799–808, Mar. 2016.
[116] Q. Chen, C. Liang, X. Wang, J. He, Y. Li, and Z. Liu, “An albumin-based theranostic nano-agent for dual-modal imaging guided photothermal therapy to inhibit lymphatic metastasis of cancer post surgery,” Biomaterials, vol. 35, no. 34, pp. 9355–9362, Nov. 2014.
[117] Q. Chen et al., “Intelligent Albumin-MnO 2 Nanoparticles as pH-/H 2 O 2 -Responsive Dissociable Nanocarriers to Modulate Tumor Hypoxia for Effective Combination Therapy,” Adv. Mater., vol. 28, no. 33, pp. 7129–7136, Sep. 2016.
[118] G. Hartung et al., “Phase I Trial of Methotrexate-Albumin in a Weekly Intravenous Bolus Regimen in Cancer Patients Phase I Trial of Methotrexate-Albumin in a Weekly Intravenous Bolus Regimen in Cancer Patients 1,” vol. 5, no. April, pp. 753–759, 1999.
[119] A. Vis, A. Van Der Gaast, B. Van Rhijn, T. Catsburg, C. Schmidt, and G. Mickisch, “A phase II trial of methotrexate-human serum albumin (MTX-HSA) in patients with metastatic renal cell carcinoma who progressed under immunotherapy,” Cancer Chemother. Pharmacol., vol. 49, no. 4, pp. 342–345, 2002.
[120] C. Bolling et al., “Phase II study of MTX-HSA in combination with Cisplatin as first line treatment in patients with advanced or metastatic transitional cell carcinoma,” Invest. New Drugs, vol. 24, no. 6, pp. 521–527, 2006.
[121] C. Unger et al., “Phase I and pharmacokinetic study of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin,” Clin. Cancer Res., vol. 13, no. 16, pp. 4858–4866, 2007.
[122] S. P. Chawla et al., “First-line aldoxorubicin vs doxorubicin in metastatic or locally advanced unresectable soft-tissue sarcoma,” JAMA Oncol., vol. 1, no. 9, pp. 1272–1280, 2015.
[123] F. Kratz, “A clinical update of using albumin as a drug vehicle – A commentary,” Journal of Controlled Release, vol. 190. 2014.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Cancer"
Cancer is a disease in which cells grow or reproduce abnormally or uncontrollably. Cancerous cells have the potential to spread to other areas of the body in a process called metastasis.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: