Assessing and Predicting Bio-environmental Interactions in Freshwater Urban Environments
Info: 8211 words (33 pages) Dissertation
Published: 20th Feb 2025
Tagged: Environmental Science
Overview
This project focuses on assessing and predicting biological and environmental interactions in freshwater urban biotas. Target environments, located in the Chattanooga area are known to be exposed to different levels of anthropogenic activities and waters are characterized by specific ecological settings.
The project will consider systematic data publicly available as well as integrate additional measurements of water quality and remote sensing. Concurrently, biodiversity will be estimated using a combination of metabarcoding, morphological, phylogenetic, and DNA taxonomy data. Goals are:
(1) Track environmental and human drivers that can cause biodiversity shifting in our focal ecosystems. This will be accomplished through continuing measurements of climate, water chemistry, human activities, and biodiversity evaluations.
(2) Investigate patterns and processes in biological diversity using rotifers as model organisms; the integration of multiple taxonomic approaches, genetic sequencing, as well as the diversity of their associated microbiome will be assessed integrating morphological and molecular tools. Phylogenetic and statistical comparative methods will be used to identify phylogenetic signal in ecological traits.
(3) Identify relationships between drivers and response variables, which can be used to predict the effects of future changes in microscopic communities. This will be accomplished through a combination of simulation, statistical, and mathematical modeling.
Intellectual Merit
Sequencing of environmental DNA is an emergent monitoring tool that promises to facilitate the accurate and cost-effective detection of species in environmental samples. However, data obtained with this technology are not routinely used to test ecosystem models that explore biodiversity shifting. This project aims to develop simulation models based on genetic data obtained from freshwater ecosystems to predict the shifting of biodiversity under disturbances. Ecologists have a long-standing interest in disturbance, but different studies define disturbance in different ways, and furthermore, it is rare to have a comprehensive understanding of multiple disturbance types across abiotic gradients.
Moreover, different studies are mostly focused on the biodiversity investigation of only one or two communities. The research proposed here is designed to systematically characterize perturbation patterns and to evaluate disturbance responses with a standardized suite of multiple biological communities of microscopic organisms, species, populations, and ecosystem variables. In the long-term, information on spatial and temporal patterns in perturbations, along with observed disturbance effects, will be combined to produce a landscape-scale synthesis of disturbance responses in freshwater ecosystems and their relationship with external drivers.
Broader Impacts
This study contributes to foster partnerships between UTC and the local community by disseminating the understanding of aquatic ecosystems with citizen scientists and the general public. Data obtained from this project will be deposited into a public repository. Further, final mathematical and statistical models, calibrated on our data, will be used to develop a user-friendly, interactive, and updateable website that allows external users to easily analyze and visualize shifting of biodiversity in freshwater ecosystems under stressors.
Moreover, the project will contribute to continuing the launch of a yearly quantitative and computational workshop, open to both local and external attendees, which has been promoted and organized by the PI starting in Spring 2019. The Biology, Geology & Environmental Science Department at UTC is in the process of offering pathways for Environmental Science majors to earn the Rivers Studies and Leadership Certificate (RSLC).
This project will provide research experiences to students that will help them to meet this requirement and earn the RSLC. Lastly, the nature of this project allows the establishment of interdisciplinary research opportunities spanning quantitative methods in biology. The funded proposal will support scholarships for undergraduate and graduate students focusing on topics in the field of computational and quantitative biology, an overlooked subject in the CoPIs Institutions.
1. Project Description
1.1 Objectives
The goal of the proposed research is to assess and predict patterns and processes in the biological diversity of microscopic species across freshwater environments that are under stressors. This work focuses on multiple types of disturbances as well as multiple biological communities and aims to combine and integrate metabarcoding, morphology, phylogenetics, and DNA taxonomy to assess the characteristics of biodiversity. The focal area includes sites important ecologically, nevertheless poorly investigated, located in the Southeastern United States (Chattanooga, Tennessee), such as the Tennessee River, its tributaries, and connected ponds and wetlands.
The Southeastern United States, is a global hotspot of freshwater biodiversity, supporting almost two-thirds of the country’s fish species, over 90% of the US total species of mussels and nearly half of the global total for crayfish species. More than a quarter of this region’s species are found nowhere else in the world. Unfortunately, this region is also a hotspot for imperilment. The number of imperiled freshwater fish species in the Southeast has risen 125% in the past 20 years, in part because recent intensive human development of this region is coupled with a low priority for conservation (Jenkins et al. 2015).
The Tennessee River, specifically, is home to more aquatic species than any other region in North America and contains one of the most diverse aquatic ecosystems in the world. Scientific research has extensively documented the causes of species imperilment in the Tennessee River, yet efforts to reverse these trends have been hampered by limited funding and lack of public awareness.
Also, most of the biodiversity studies in the Southeastern US have focused on the observation of large invertebrates or fish, whereas little is known about the microscopic communities. Microscopic organisms are abundant and ubiquitous in freshwater ecosystems, performing key functions such as nutrient cycling and sediment stability. Yet, their unexplored diversity and response to disturbances represent one of the major challenges in biology and currently limits our capacity to understand, mitigate, and remediate the consequences of pollution and environmental change.
The proposed research aims to address fundamental questions about biodiversity assessment and biodiversity shifting of microscopic organisms in freshwater environments, such as:
What organisms live there?
Why are they found here?
In particular, an organism’s genetics, morphology and the surrounding ecology contribute to their ability to thrive in a certain habitat. How are these organisms impacted by a changing environment?
How does biodiversity assemblage change across environments?
What are the biological and ecological correlations among and within freshwater microscopic communities?
Can we predict biodiversity shifting with future environmental changes?
To this end, the team will employ a novel workflow that systematically collects several ecological parameters from each sampled location and remote-sensing data. Concurrently, the biodiversity of large communities, including Archea, Bacteria, fungi, protists, and small invertebrates will be investigated using metabarcoding.
Rotifers will be used as model organisms to assess patterns of diversity and distribution in populations of microscopic metazoans. A combination of morphological, phylogenetics, and DNA taxonomy will be performed to single individuals of rotifers across populations. Comparative phylogenetic, statistical, and mathematical models will be developed to encapsulate biodiversity shifting in changing environments at both a community and population level. The specific objectives of this proposal are to:
- Disentangle the biodiversity of large communities of microscopic organisms. Multiple-primer metabarcoding approaches will disclose the taxonomic diversity of prokaryotes (Archaea and Bacteria), fungi, protists, and small invertebrates. DNA taxonomy will be applied on single rotifer individuals. Two publications are anticipated.
- Disclose ecological interactions among major taxa. We predict that community structure changes are affected by environmental conditions as well as pairwise/multiple interactions among species. Statistical models will test interactions across taxa and communities as well as correlations between biological diversity changes and disturbances. Two publications are anticipated.
- Characterize patterns of diversity and distribution of rotifers. An integrative taxonomic approach will be used to identify species, characterize the rotifer fauna of freshwater ecosystems, and aid future researchers in identifying specimens by producing a library of DNA barcodes from expert-identified specimens. Two publications are anticipated.
- Understand patterns and processes in biological diversity using rotifers as organism models by applying comparative phylogenetic and statistical models. Two publications are anticipated.
- Develop mathematical and statistical models to simulate and predict stressor scenarios for projected future land uses. Two publications are anticipated.
- Employ the developed mathematical and statistical models to reconstruct a software available to the general public aimed at predicting biodiversity by changing of abiotic parameters. One publication is anticipated.
The proposed activities will enable the team to build a firm foundation for a lifetime of quality research and teaching. The team is committed to excellence in education as demonstrated by their strong teaching evaluations, mentoring, and outreach activities (see BioSketch). This research will directly integrate several educational objectives including
i) mentoring of 3 graduate students, and 5-7 undergraduate researchers per semester,
ii) hosting computational and quantitative training workshops led by world expert mathematicians, statisticians, and computational biologists,
iii) enhancing the PI’s existing courses, and
iv) developing an open-access software for external users aimed at visualize biological response to stressors.
Knowledge gained from assessments will be used to dynamically improve approaches, maximizing impact and increasing retention of researchers in the field.
The CoPIs are uniquely qualified and well-positioned for this work. The PI has been working with microscopic aquatic organisms for over ten years (REFERENCES), is an expert in biodiversity estimates using different morphological, metabarcoding, DNA taxonomy, and phylogenetic tools (REF), and collaborates with world experts on environmental DNA analyses and rotifers. The CoPIs have been working on developing models to understand environmental changes, the impact of human activities, conservation, biological processing, and ecological correlations (reference).
All facilities, equipment, and resources needed to conduct this research are available to the PI and CoPIs (see Facilities, Equipment, and Other Resources statement).
1.2 Background and Significance
For freshwater ecosystems, biodiversity provides essential ecosystem services such as fisheries, provision of water for drinking and food production, as well as transportation and recreational purposes (Balian et al., 2008). Thus, the maintenance of this remarkable variety of life is one of the important keys to the retention of these services, which are essential to human health and well-being (Corvalan et al., 2005). Although freshwater ecosystems cover only 0.8% of the planets’ surface, they account for more than 10% of all animal species.
The Southeastern United States is a global hotspot of freshwater biodiversity but, as generally around the world, also of endangerment including habitat destruction and fragmentation, the introduction of invasive species, pollution, human population growth as well as overharvesting. Nonetheless, while most protected lands are in the West, the vulnerable species are largely in the Southeast. The decline in freshwater biodiversity is exceeding that of other systems (Dudgeon et al., 2006) and, despite knowing about biodiversity’s importance for a long time, human pressures on freshwater ecosystems have increased tremendously over the last century.
Investigating freshwater biodiversity in urban areas, such as in Chattanooga, is particularly important to develop conservation programs (Fig. 1).
Cities are investing billions of dollars in climate change adaptation to combat the effects of sea‐level rise, temperature extremes, increasingly intense storm events, flooding, and water scarcity. Natural ecosystems have enormous potential to contribute to city resilience, and so, actions that rely on this approach could sustain considerable co‐benefits for biodiversity. As suggested by Butt et al (2018) planners and city governments should incorporate biological conservation into climate adaption plans, for the mutual benefit of urban societies and their biodiversity. However, it is rare to have a comprehensive understanding of multiple disturbance types across abiotic gradients. Moreover, among freshwater organisms, only fish and mollusk are routinely investigated in this area whereas the diversity and distribution of the other organisms, especially the small species, is largely underestimated.
Recent advances in DNA sequencing technology enables large-scale assessments of biodiversity of microscopic species implemented at the community level. Specifically, metabarcoding is a cost-effective approach to assess biodiversity of broader communities and interactions of taxa across trophic levels and to appraise temporal ecosystem variations following natural or anthropogenic events (reference). DNA amplification, purification, and sequencing for metabarcoding can be conducted using standard protocols and designed primer sequences available from literature (reference:[27], [28]. Certain biases, mostly related to the use of universal primers, are known to affect the results obtained with metabarcoding [29].
To limit such biases, it has been suggested to apply a metabarcoding with the use of multiple taxon-specific primers [30]. This would highlight the diversity of several microscopic components, such as Bacteria, Archea, protists, fungi, as well as the usually neglected invertebrates smaller than
Among meiofaunal organisms, monogonont rotifers are ideal models to study micro-evolutionary adaptation and its eco-evolutionary consequences. Rotifers are relatively small and easy to handle and their short generation time allows for rapid evolutionary trait responses. Importantly, two additional features associated distinguish them from many other metazoans and make them especially amenable for work on rapid evolution: i.e., the alternation of clonal with sexual reproduction and the tight link between sexual reproduction and the formation of diapausing eggs. With diapausing eggs, populations can survive adverse conditions and disperse both in space and time; these eggs can be used to reconstruct evolutionary trajectories of natural populations through time via the ‘resurrection’ of genotypes that may be recovered from stratified lake sediments [21]–[25]. Patterns of diversity in monogonont rotifers have been investigated across several species, mostly within the genus Brachionus (Fig. 2).
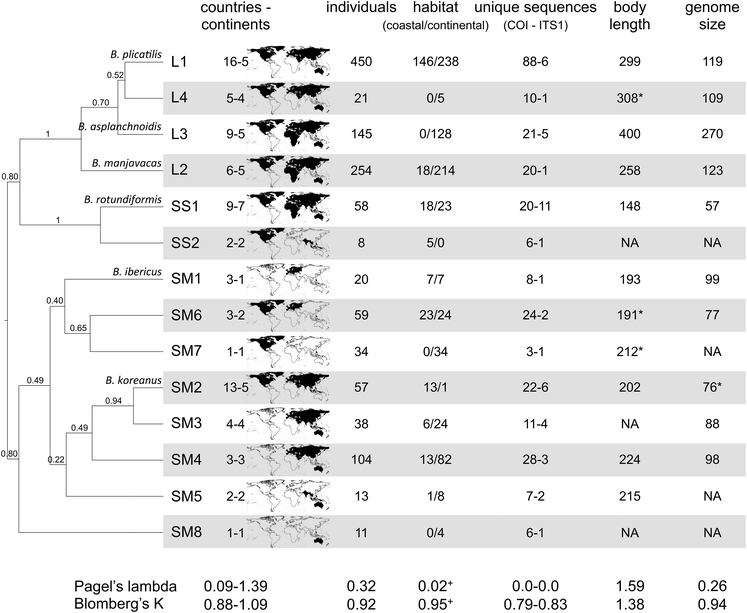
However, the ecological measurements at a local scale have been rarely considered to understand patterns and processes in biological diversity, which is a critical task given current and rapid environmental change.
Understanding interactions between microbes and invertebrates can lead to insights of broader relevance for the ecosystem and ecosystem health. Besides microbes present in the external environment, research efforts have focused on microbial associations known to be critical for organismal health or defense (e.g., the human microbiome [16], [17]) as well as in-depth studies of well-characterized symbionts that have co-evolved with hosts (e.g., sponge and coral reef microbiomes [18], [19]). Animals evolve in a world teeming with microbes, which play pivotal roles in their health, development, and evolution. Life as we know it would not exist without the profound impact of beneficial host-microbe interactions. Studies of host-associated microbes are critical for advancing our understanding of ecology and evolution across diverse taxa and ecosystems. A range of immune components are conserved across almost all animal life, and functional roles may be conserved in invertebrates. While emerging evidence from invertebrate taxa has underlined the evolutionary and ecological significance of microbiome assemblages, only a few studies considered the interaction between microbes and meiofauna (Fig. 3), and none on rotifers.
In conclusion, biological community assemblages are diverse, maximizing opportunities for species-specific responses to individual components of contamination, and community changes are highly specific to the type and severity of contamination, as well as the interaction of the two. Areas with high levels of species and genetic diversity are likely to have a more complex ecosystem, with a variety of food webs and biotic interactions. Multifactoriality is expected to play a pivotal role, therefore, many studies are currently focused on collecting information on the biodiversity and environmental factors that potentially influence ecosystem health [14].
Modeling of ecological processes is not a recent area of scientific research but it is currently evolving fast due to the technological advances in computing power [12]. Mathematical models can be readily used as tools for the efficient assessment of environmental quality, for studying the ecosystem functioning properties, and for monitoring biodiversity in complex biota [13]. Simulations using such large biologically realistic data sets with environmental interactions that influence the risk of a complex ecosystem are a convenient and useful way to assess the performance of statistical methods. A variety of different mathematical models to explain biodiversity exists at the moment, each one with its advantages and disadvantages. However, there is still a lack of efficient and thoroughly tested statistical models that can be used to identify implicated features and their interactions. Moreover, environmental DNA data are not routinely used to test ecosystem models that explore biodiversity shifting under disturbance [10].
1.3 Research Plan and Preliminary Results
The proposed research seeks to characterize the biodiversity in the Southeastern urban freshwaters, explore the nonlinear multispecies response to environmental gradients and predict spatial and temporal changes of biodiversity under disturbance. To this end, the PI and her team will:
1) collect samples from sites diverse in terms of human population/urbanization and land usage,
2) characterize genetic diversity of the microscopic biological entities present in the environment using metabarcoding,
3) investigate patterns of diversity and distribution of rotifers integrating morphological, phylogenetic, and DNA taxonomy approaches,
4) develop simulation models to predict biodiversity shifting with future environmental changes.
All of the proposed research activities will directly incorporate training of young scientists and foster career development for the PIs, establishing a firm foundation for a lifetime of contributions to freshwater biodiversity and ecology research and education (See Long-Term Vision).
The research team will consist of the PI and three CoPIs (Lakmali Weraseena, Eric Laflamme, and Nyssa Hunt), one Ph.D. student in mathematics (TBD) and two M.S. students in Environmental Science (Adrianna Hodges, TBD), and a team of undergraduates. The PI will organize the sampling and genetic analyses (Objective 1), train students in environmental science, conduct taxonomic work (Objective 2 in part), and oversee all aspects of the project. The CoPIs will write manuscripts, present at meetings, conduct outreach. Lakmali Weraseena and Eric Laflamme will perform the statistical analyses, develop mathematical and statistical models, and construct a website open to external users aimed at estimating and visualizing biodiversity shifting in changing environments.
Nyssa Hunt will perform remote sensing analyzing and map visualization with GIS. The graduate students will be encouraged to explore their own questions within the scope of this proposal but will be expected to become expert in quantitative biology with three dissertation chapters focused on biodiversity estimates and modeling in the Southeast US. With the help from undergraduates and the PI, they will lead the remaining tasks of Objective 2 (see below). Undergraduates will use metabarcoding, light microscopy, phylogenetics, and DNA taxonomy to characterize diversity, identify rotifer specimens, and produce a reference DNA barcode database.
Objective 1 – Sampling and Environmental Parameters Investigation
This project is focused on 12 sites located in Chattanooga, Tennessee, most of them are historically investigated by the Tennessee Department of Environment and Conservation (TDEC). Preliminary analyses of parameters measured by TDEC show significant differences of values across stations, showing that parameters known to decrease biodiversity are higher in south Chattanooga,. certain parameters fundamental to set up the model are available, including chemical contaminants, E. colim temperature, turbidity. There are two classifications of pollutants that affect water bodies in Chattanooga.
Pollutants such as pesticides, herbicides, detergents, and fecal matter that are carried by storm water runoff into creeks and streams are considered non-point source pollution, since they come from various places and the source cannot be quickly identified. If the source was easily identifiable, such as a pipe discharging or smoke stack, they would be considered point source pollution. The two main pollutants currently affecting Chattanooga’s watersheds are Escherichia coli and suspended solids such as sediment and industrial waste products (reference). Parameters will be measured with …

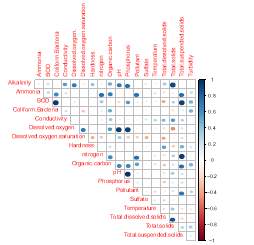
Therefore, we expect differences in biodiversity among them. From each of the three locations, water, sediment, and algae/plants will be sampled, in three replicates for each site, in order to assess benthic, planktic, and epiphytic biodiversity, respectively. For each sample, environmental parameters (i.e., nutrients, turbidity, pH, conductivity, dissolved oxygen, and nitrates) will be measured. Sampling and assessment iof environmental parameters will be conducted bi-monthly. Samples will be immediately transported to the laboratory at the University of Tennessee in Chattanooga to be processed for biodiversity assessment.
Objective 2 – Community Biodiversity Assessment
Preliminary observation of samples collected from such location show a higher biodiversity than expected. Even the most pollutant area, show #rotifer species, etc..chironomid larvae and different community composition with different nematode/copepod ration. Etc. This approach consists of obtaining genetic information of all biological entities present in the environment. DNA will be extracted from the samples using commercial kits such as PowerSoil and PowerWater DNA isolation kits (Mobio, Carlsbard, CA) according to the manufacture’s protocol. DNA amplification, purification, and sequencing for metabarcoding will be conducted using standard protocols and designed primer sequences available from literature [27], [28].
Certain biases, mostly related to the use of universal primers, are known to affect the results obtained with metabarcoding [29]. To limit such biases, we plan to apply a metabarcoding approach with the use of multiple taxon-specific primers [30]. Specifically, we plan to assess diversity by investigating the mitochondrial gene Cytochrome Oxidase Subunit I (COI), as well as the small ribosomal subunit (rRNA 18S) and Internal Transcribed Spacer (ITS) genes for eukaryotes, and the 16S rRNA gene for prokaryotes. Moreover, as a recent publication suggested [31], we will consider the results of the eukaryote mitochondrial COI and prokaryote 16S rRNA amplicons to estimate biomass of species, besides taxonomic diversity.
To reconstruct Operational Taxonomic Units and Sequence Variants, which delimit taxa in the sample, QIIME2 will be applied [32]. Moreover, we plan to apply Keck’s method [33], a bioinformatics software package designed to predict metagenome functions in organisms from marker genes. A common observation is that continuous traits of closely related species in a phylogeny are often similar, especially when traits are under selection pressure of the environment. Therefore, this method, as well as PICRUSt [34], relies on the assumption that phylogeny and function are sufficiently linked to make predictions about a community’s functional potential.
While PICRUSt is restricted to the 16S rRNA gene, Keck’s method can be extended to any known marker genes depending on which group of organisms one wishes to study. This method can be considered taxonomy-free because instead of blasting short reads against a database of reference taxa, it uses phylogenetic information. Phylogenetic distances, such as UniFrac, which is widely used in microbial ecology and has been adopted in metabarcoding, can greatly improve the accuracy of community similarity and dissimilarity matrices [35]. Biodiversity will be measured as richness (number of species and abundance), species composition in the community, and phylogenetic diversity.
Objective 3 – Processes in Rotifer Biodiversity
The goal is discovering the genetic basis behind adaptive phenotypes [26]. I plan to collate a dataset of previously available and newly generated sequences of COI and ITS1 for isolates of the Brachionus complex and apply three approaches in DNA taxonomy (i.e. ABGD, PTP, and GMYC) to identify and provide support for the existence of cryptic species within the complex. I plan to use these results to explore phylogenetic signal in morphometric and ecological traits, and to understand correlation among the traits using phylogenetic comparative models.
Objective 4 - Microbiome associate
I plan to investigate the microbiome associate with rotifer species using a metabarcoding approach. I expect to discover assemblage patterns of rotifer microbiota that show a correlation with the phylogeny of the host, core microbiota, or are correlated to environmental parameters, accessory microbiota. Genetic and genomic approaches will be used to investigate patterns of diversity of rotifers, explore phylogenetic signals in morphometric and ecological traits, and understand correlations among the traits using phylogenetic comparative models.
Objective 5 - Statistical Approach
We predict that community structure changes are affected by environmental conditions as well as pairwise/multiple interactions among species. These interactions can be trophic or symbiotic. We aim to test interactions across taxa and communities as well as the correlations between biological diversity changes and disturbances. We expect that interaction strength between biological diversity and ecological conditions may decrease with the number of links between species, damping the effects of any disturbance.
Statistical Modeling Framework. We will consider time-series data on biodiversity response measure, such as species abundance, that span over
n+1
time steps (e.g., years) and involve
m
species. The standard first order multivariate autoregressive in vector form can be defined by
yt
=c +
Ayt–1
+
et
Where
yt
(may need to be log transformed) denotes the abundance of all species at time
t
and c is a
m×1
vectorof intercepts of all species. And the interaction matrix
A
is a
m×m
matrix. The noise (error) term
e
is assumed to follow the multivariate normal distribution
et~N(0, Ω)
where
Ω
is the variance-covariance symmetric matrix with dimension of
m×m
This model can also be generalized to other data distribution by letting
yt
be the linear predictor within a generalized linear modeling framework dependent on the distribution of the data to be collected. Random factors can be implemented through a latent factor approach.
Model Fitting. We will parameterize the model in a Bayesian framework. This framework enables one not only to parameterize the model described above but also to extend it to involve environmental covariates, species traits, phylogenetic relationships, as well as e.g. a spatially hierarchical or a spatially explicit study design. Further, in addition to normally distributed data, it includes as data models Bernoulli distribution (with probit link-function) for presence– absence data and Poisson and over-dispersed Poisson distributions (with log link-function) for count data.
To evaluate the performance of the above described statistical model, we may define a set of alternative models (for instance, no interspecific interactions, full interaction, and sparse interactions). We will generate simulated data using Monte Carlo Method based on different assumptions of the models.
Alternatively, if time series data is not available, we will consider nested ANOVA models. We will test for covariate effects by comparing full and reduced models with F-tests.
Moreover, interaction networks may be built and structured by species traits if possible. Incorporating other factors into the modeling framework may further improve our upstanding of the interaction between biodiversity and change of community and environment.
Statistical Model Development (6th-12th month PI, CoPIs #1, 2, 3, students a, b, c)
Environmental parameters and biological data will be combined in the validated model.Results will then be used to predict the pollutants loading at different spatial locations along the creek as well temporally with changing land uses over the time. In addition, the model will be used to create pollutants stressor scenarios for projected future land uses of the watershed.
The last three months of the proposed project will be also devoted to preparing manuscripts for submission, present results during conferences, and develop projects suitable for extra-mural funding agencies.
Future perspectives
Simulations using large biologically realistic data sets with known gene-gene and gene-environment interactions that influence the risk of a complex ecosystem are a convenient and useful way to assess the performance of statistical methods. Nonetheless, in a recent review, Donohue et al. [36] made several important points regarding the limitations of previous research: 1) theoretical studies tend to focus on the effects of a single pulse perturbation; 2) few studies combine theory and empirical measurements; 3) most empirical studies focus on population or community characteristics rather than ecosystem functions and processes; and 4) there is a strong bias (> 50%) towards non-aquatic systems. This topic is particularly salient not only because many elements of global change show an increase in both mean and variance over time, but also because the threshold for a state change may change with different underlying conditions [37].
Results of this project will provide the basis to develop further simulation models applicable to all freshwater ecosystems. Importantly, aliquots of DNA obtained during this project will be preserved for a metagenomics approach. The application of metagenomics provides access to the functional gene composition of microbial communities and thus gives a much broader description than phylogenetic surveys, which are often based only on the diversity of one or a few genes [38]. On its own, metagenomics gives genetic information on potentially novel biocatalysts or enzymes, genomic linkages between function and phylogeny for uncultured organisms, and evolutionary profiles of community function and structure in a “system ecology” perspective. A system ecology approach is increasingly relevant in light of the replenishing “microbial seed bank” that appears to persist in the ecosystems [32], [39] and mounting evidence that functional genes are a more important factor for community assembly versus species themselves [40].
Moreover, statistical analyses provided by the present study will be fundamental to develop mathematical models of ecological networks [41]. Ecological networks describe and compare the interactions between the biotic and abiotic elements of real ecosystems, for example, a food web is a network capturing the trophic interactions between the various species.
Recent advances in DNA sequencing techniques (metabarcoding and metagenomics) allow researchers to construct and describe such networks in unprecedented levels of detail and simulate realistic network responses to external perturbations.
Broader Impact
Although the protection and conservation of freshwaters have fallen behind the conservation of terrestrial ecosystems, there is an ongoing attempt to map global freshwater biodiversity hotspots. Interest in restoration or rehabilitation of freshwater ecosystems has largely increased (e.g. Hart & Poff, 2002) and several restoration programs are well developed and partly successfully practiced (Downes et al., 2002). Human impact represented by climate change, land use, and use of plastics and microplastics affect a large proportion of freshwater ecosystems and reservoirs worldwide [42], [43]. Rivers, lakes, and wetlands are under intense pressure from overuse, pollution, and habitat degradation [44]. The services that aquatic ecosystems can provide to society have been greatly reduced, and the biota has been strongly affected, with several aquatic species disappearing from entire ecoregions.
Public health risks associated with polluted water include gastroenteritis, otitis, conjunctivitis, and skin infections [45], besides increased carcinogenesis, immunodeficiency, lower cognitive performance, and other behavioral deficits [46].
Environmental health risk associated with polluted water includes a decrease of ecosystems and recreational services as well as drastic reduction in biodiversity [47]. That said, the effects of exposures to mixtures of pollutants on the microbiome, and in turn animal physiology and viability, are poorly understood. Because the analysis of complex biotas is a fundamental topic in management, monitoring, and conservation of the environment, which directly impacts human well-being, we plan to share our understanding of interactions in aquatic ecosystems with teachers and students, local environmental managers, citizen scientists, and the general public.
At the University-level, this project aims to advance the strategic plans of UTC and the College of Science by nurturing and empowering scholarship, discovery, and entrepreneurial initiatives; including theoretical and applied activities that routinely engage students, faculty, and community partners; and providing interdisciplinary research opportunities. That is, this project helps establish interdisciplinary and long-term collaborations across departments. The program will provide research opportunities for a diverse group of undergraduates and interdepartmental courses for students at different levels. Specifically, in addition to offering scholarships for three graduate students as described above, this project will strongly contribute to new Environmental Science majors to earn the Rivers Studies and Leadership Certificate (RSLC). The program is expected to begin in Fall 2019. One requirement of the program is that students complete a professional experience (i.e., internship, research, etc.). This project will provide research experiences to students that will help them to meet this requirement and earn the RSLC.
REFERENCES
[1] L. Peters, H. Hillebrand, and W. Traunspurger, “Spatial variation of grazer effects on epilithic meiofauna and algae,” J. North Am. Benthol. Soc., vol. 26, no. 1, pp. 78–91, 2007.
[2] P. D. P. C. et al., “Cross‐system comparisons elucidate disturbance complexities and generalities,” Ecosphere, vol. 2, no. 7, p. art81.
[3] T. M. G., “Disturbance and landscape dynamics in a changing world1,” Ecology, vol. 91, no. 10, pp. 2833–2849.
[4] M. F. Mare, “A study of a marine benthic community with special reference to the micro-organisms,” J. Mar. Biol. Assoc. United Kingdom, vol. 25, no. 3, pp. 517–554, 1942.
[5] F. Leasi, C. Gaynus, A. Mahardini, T. N. Moore, J. L. Norenburg, and P. H. Barber, “Spatial and ecologic distribution of neglected microinvertebrate communities across endangered ecosystems: meiofauna in Bali (Indonesia),” Mar. Ecol., vol. 37, no. 5, 2016.
[6] L. C. de Faria et al., “The use of metabarcoding for meiofauna ecological patterns assessment,” Mar. Environ. Res., vol. 140, pp. 160–168, 2018.
[7] C. Wurzbacher et al., “DNA metabarcoding of unfractionated water samples relates phyto-, zoo- and bacterioplankton dynamics and reveals a single-taxon bacterial bloom,” Environ. Microbiol. Rep., vol. 9, no. 4, pp. 383–388.
[8] A. Valentini et al., “Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding,” Mol. Ecol., vol. 25, no. 4, pp. 929–942, 2016.
[9] P. Taberlet et al., “Soil sampling and isolation of extracellular DNA from large amount of starting material suitable for metabarcoding studies,” Mol. Ecol., vol. 21, no. 8, pp. 1816–1820, 2012.
[10] V. J. Coles et al., “Ocean biogeochemistry modeled with emergent trait-based genomics,” Science (80-. )., vol. 358, no. 6367, p. 1149 LP-1154, Dec. 2017.
[11] M. Brinke et al., “Using meiofauna to assess pollutants in freshwater sediments: A microcosm study with cadmium,” Environ. Toxicol. Chem., vol. 30, no. 2, pp. 427–438.
[12] H. H. Shugart et al., “Computer and remote-sensing infrastructure to enhance large-scale testing of individual-based forest models,” Front. Ecol. Environ., vol. 13, no. 9, pp. 503–511.
[13] A. Ostmann and P. Martínez Arbizu, “Predictive models using randomForest regression for distribution patterns of meiofauna in Icelandic waters,” Mar. Biodivers., vol. 48, no. 2, pp. 719–735, 2018.
[14] P. A. Sandifer, A. E. Sutton-Grier, and B. P. Ward, “Exploring connections among nature, biodiversity, ecosystem services, and human health and well-being: Opportunities to enhance health and biodiversity conservation,” Ecosyst. Serv., vol. 12, pp. 1–15, 2015.
[15] C. Roland Pitcher et al., “Exploring the role of environmental variables in shaping patterns of seabed biodiversity composition in regional-scale ecosystems,” J. Appl. Ecol., vol. 49, no. 3, pp. 670–679.
[16] N. N. Schommer and R. L. Gallo, “Structure and function of the human skin microbiome,” Trends Microbiol., vol. 21, no. 12, pp. 660–668, 2013.
[17] J. Walter and R. Ley, “The Human Gut Microbiome: Ecology and Recent Evolutionary Changes,” Annu. Rev. Microbiol., vol. 65, no. 1, pp. 411–429, 2011.
[18] D. G. Bourne, K. M. Morrow, and N. S. Webster, “Insights into the Coral Microbiome: Underpinning the Health and Resilience of Reef Ecosystems,” Annu. Rev. Microbiol., vol. 70, no. 1, pp. 317–340, 2016.
[19] U. Hentschel, J. Piel, S. M. Degnan, and M. W. Taylor, “Genomic insights into the marine sponge microbiome,” Nat. Rev. Microbiol., vol. 10, p. 641, Jul. 2012.
[20] J. P. D., R. ASHOK, P. SUSANNE, L. J. A., S. R. W., and C. J. K., “How do consumers deal with stoichiometric constraints? Lessons from functional genomics using Daphnia pulex,” Mol. Ecol., vol. 20, no. 11, pp. 2341–2352.
[21] K. W. Charles, R. J. A., and W. L. J., “A new approach to historical reconstruction: Combining descriptive and experimental paleolimnology,” Limnol. Oceanogr., vol. 44, no. 5, pp. 1232–1247.
[22] E. Decaestecker et al., “Host–parasite ‘Red Queen’ dynamics archived in pond sediment,” Nature, vol. 450, p. 870, Nov. 2007.
[23] F. Dagmar et al., “A millennial‐scale chronicle of evolutionary responses to cultural eutrophication in Daphnia,” Ecol. Lett., vol. 17, no. 3, pp. 360–368.
[24] S. A. J. Declerck and S. Papakostas, “Monogonont rotifers as model systems for the study of micro-evolutionary adaptation and its eco-evolutionary implications,” Hydrobiologia, vol. 796, no. 1, pp. 131–144, Jul. 2017.
[25] S. Mills et al., “Fifteen species in one: deciphering the Brachionus plicatilis species complex (Rotifera, Monogononta) through DNA taxonomy,” Hydrobiologia, 2016.
[26] C. E. Ellison et al., “Population genomics and local adaptation in wild isolates of a model microbial eukaryote,” Proc. Natl. Acad. Sci., vol. 108, no. 7, p. 2831 LP-2836, Feb. 2011.
[27] J. G. Caporaso et al., “Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms,” ISME J., vol. 6, no. 8, pp. 1621–1624, Aug. 2012.
[28] D. W. Yu et al., “Biodiversity soup: Metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring,” Methods Ecol. Evol., vol. 3, no. 4, pp. 613–623, 2012.
[29] F. Leasi et al., “Biodiversity estimates and ecological interpretations of meiofaunal communities are biased by the taxonomic approach,” Commun. Biol., vol. 1, no. 1, p. 112, 2018.
[30] M. Stat et al., “Ecosystem biomonitoring with eDNA: metabarcoding across the tree of life in a tropical marine environment,” Sci. Rep., vol. 7, no. 1, p. 12240, 2017.
[31] I. Bista et al., “Performance of amplicon and shotgun sequencing for accurate biomass estimation in invertebrate community samples,” Mol. Ecol. Resour., vol. 18, no. 5, pp. 1020–1034.
[32] J. G. Caporaso et al., “QIIME allows analysis of high-throughput community sequencing data,” Nat. Methods, vol. 7, no. 5, pp. 335–336, May 2010.
[33] F. Keck, F. Rimet, A. Bouchez, and A. Franc, “phylosignal: an R package to measure, test, and explore the phylogenetic signal,” Ecol. Evol., vol. 6, no. 9, pp. 2774–2780.
[34] M. G. I. Langille et al., “Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences,” Nat. Biotechnol., vol. 31, p. 814, Aug. 2013.
[35] C. Lozupone, M. Hamady, and R. Knight, “UniFrac – An online tool for comparing microbial community diversity in a phylogenetic context,” BMC Bioinformatics, vol. 7, no. 1, p. 371, 2006.
[36] D. Ian et al., “Navigating the complexity of ecological stability,” Ecol. Lett., vol. 19, no. 9, pp. 1172–1185.
[37] M. Scheffer and S. Carpenter, “Catastrophic Regime Shifts in Ecosystems: Linking Theory to Observation,” Trends Ecol. Evol., vol. 18, pp. 648–656, 2003.
[38] V. J. Coles et al., “Ocean biogeochemistry modeled with emergent trait-based genomics,” Science (80-. )., vol. 358, no. 6367, p. 1149 LP-1154, Dec. 2017.
[39] J. G. Caporaso et al., “Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms,” ISME J., vol. 6, no. 8, pp. 1621–1624, Aug. 2012.
[40] C. Burke, P. Steinberg, D. Rusch, S. Kjelleberg, and T. Thomas, “Bacterial community assembly based on functional genes rather than species,” Proc. Natl. Acad. Sci., vol. 108, no. 34, p. 14288 LP-14293, Aug. 2011.
[41] Z. D. Kurtz, C. L. Müller, E. R. Miraldi, D. R. Littman, M. J. Blaser, and R. A. Bonneau, “Sparse and Compositionally Robust Inference of Microbial Ecological Networks,” PLOS Comput. Biol., vol. 11, no. 5, pp. 1–25, 2015.
[42] B. W. Brooks et al., “Are harmful algal blooms becoming the greatest inland water quality threat to public health and aquatic ecosystems?,” Environ. Toxicol. Chem., vol. 35, no. 1, pp. 6–13.
[43] Y. Lu et al., “Impacts of soil and water pollution on food safety and health risks in China,” Environ. Int., vol. 77, pp. 5–15, 2015.
[44] A. Binzer, C. Guill, B. C. Rall, and U. Brose, “Interactive effects of warming, eutrophication and size structure: impacts on biodiversity and food-web structure,” Glob. Chang. Biol., vol. 22, no. 1, pp. 220–227, 2016.
[45] E. Dewailly, C. Poirier, and F. M. Meyer, “Health hazards associated with windsurfing on polluted water.,” Am. J. Public Health, vol. 76, no. 6, pp. 690–691, Jun. 1986.
[46] R. D. Masters and M. J. Coplan, “Water treatment with silicofluorides and lead toxicity,” Int. J. Environ. Stud., vol. 56, no. 4, pp. 435–449, 1999.
[47] H. M. Pereira et al., “Essential Biodiversity Variables,” Science (80-. )., vol. 339, no. 6117, p. 277 LP-278, Jan. 2013.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Environmental Science"
Environmental science is an interdisciplinary field focused on the study of the physical, chemical, and biological conditions of the environment and environmental effects on organisms, and solutions to environmental issues.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: