Clinical Data Management Quality System Components
Info: 9084 words (36 pages) Dissertation
Published: 24th Jan 2022
Tagged: Information Systems
1. INTRODUCTION
Clinical trials are conducted to gather the data essential to deliver information for industry, regulators and academia to make decisions about the safety and efficacy of the disease, illness, or preventative medications under study. A huge quantum of clinical data is generated by these clinical trials. Quality of this clinical data is of chief importance for it determines the data reliability [1]. The collection, cleaning and maintenance of this clinical data is essential prior to processing and analysing it for effective outcome of clinical trials in compliance with the regulatory standards such as ICH-GCP [2]. As defined by “The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH)- “Good Clinical Practice (GCP) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety and well-being of trial subjects are protected, consistent with the principles that have their origin in the Declaration of Helsinki, and that the clinical trial data are credible” [3].
The GCP guidelines E6 (R1), remained unchanged for almost two decades. In November 2016, the ICH-GCP guidelines were revised to integrate the addendum E6 (R2). One of the promising addendums as identified by industry experts is the emphasis on adoption of Risk Based Monitoring (RBM)and Quality Management System (QMS). In addition, it defines standards for the management of essential documents, electronic records, and use of IT tools.
Since the previous revision of these guidelines, the environment in clinical trials has fundamentally changed. Clinical trials have grown dramatically in terms of both number and complexity. Technology has improved at a rapid pace since the issue of E6 guidelines in 1996. Internet, electronic data capture, real time clinical data review etc were merely an inception towards the improvisation of trial data management back then. The cost and complexity of trial has however, counterbalanced the technology progress. It has contributed in creating new challenges such as increased inconsistency in clinical investigator experience, treatment choice, site set-up and standard of healthcare in general [4].
As a result, the pressure on sponsors has increased while the quality standards have become firmer.
1.1 Quality Management System
The regulatory environment for conducting trials has been strict since the start. As defined by the ICH, the section 5.0 of the addendum E6 (R2) emphasis on sponsor’s responsibilities pertaining to Quality Management as:

Figure 1: Sponsor’s responsibilities pertaining to Quality Management as per ICH GCP E6 (R2)
(ICH, Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2), Prepared by the ICH E6(R2) Expert Working Group, 2017 http://www.ich.org/home.html)
This update has made it obligatory for sponsors to identify critical data and process and subsequently implement a solid system to maintain quality throughout all stages of the trial process.
1.2 Importance of Quality Management System in Clinical Data Management
Quality can be simply defined as the degree of excellence for products and services. Quality planning determines standards and scope of how output is going to be measured and analysed. The primary objective of quality management is to protect the rights safety and well-being of subjects at the same time to ensure the credibility of data for analysis. An ideal QMS should ensure compliance with the study protocol, regulatory and ethical requirements. It must analyse the data and identify areas that may require further investigation and action [5].
Since the focus is primarily on the reliability of trial results, Clinical Data Management (CDM) processes come into limelight as this domain of clinical research is responsible for collecting, maintaining and delivering trial data for statistical analysis. Clinical Data Management is a multidisciplinary area that collectively involves collection of reliable, high-quality and statistically sound data generating from the clinical trials. The platform used by clinical data managers to perform these activities is known as a Clinical Data Management System (CDMS) [6]. Huge data is collected and from the start of the trial. Thus, maintaining the data that is collected and the clinical data management system it is collected in is equally important.
1.3 Clinical Data Management Process Flow
CDM process is divided into three phases- Set-up, Conduct and Close-out.
Set-up/ Start-up phase:
This is the initial and usually the most critical phase of clinical data management. Once a protocol is approved, the clinical data management team begins with the development of clinical data management system as defined in the Data Management Plan (DMP). Majority of the software tools for CDMS employed in pharmaceutical organizations are commercial. Only a limited number of open source tools are available. The most commonly used CDM tools are ORACLE, RAVE, MACRO [1,7].
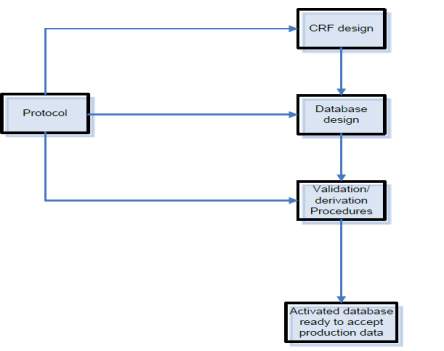
Figure 2: An overview on clinical data management set-up activities (Clinical Data Management- An introduction, QA Data, www.quadata.co.za)
The main components of this system are:
1. Case Report Form (CRF) design and development
CRF is used to capture the intended data for analysis per visit schedule as defined in the protocol. Depending on the project, these CRFs could be paper (pCRF) or electronic (eCRF). If it is paper CRF, the data is captured on paper and then transferred in the database and if it is electronic, the data is directly captured in the CDMS by the site staff. Each CRFs are assigned annotations. CRF annotation is generally defined as a blank CRF with markings (annotations) which coordinate each datapoint in the form to its corresponding dataset name. In simple language it communicates where the data collected for each question is stored in CRF. These annotations form the first step in translating the CRFs into a database application [8].

Figure 3. An example of CRF annotations (red text) for data fields related to Informed Consent and Demographics
Importance of CRF annotations and data standards
Once a trial is completed, the data is submitted to the regulatory authorities like FDA. Since regulatory bodies receive data from all over the world in different formats. Data insight often becomes a labour-intensive process as clinical data reported by individual trials are in in numerous ways. Consequently, the regulatory bodies made it mandatory to submit the clinical and non-clinical data only in a certain standard structure and tabulation as a part of a product application. These standard models are collectively known as Study Data Tabulation Model (SDTM) [9].
The Clinical Data Standards Initiative aims to streamline the process by developing Data Standards that are industry-wide in nature and are catering to priority Therapeutic Areas (TAs). This initiative is in collaboration with Critical Path Institute (C-Path), Clinical Data Interchange Standards Consortium (CDISC), National Cancer Institute- Enterprise Vocabulary Service (NCI-EVS) and FDA as a part of the Coalition For Accelerating Standards and Therapies (CFAST). These standard supports both improving patient safety and outcome and simultaneously the exchange and submission of clinical research and meta-data [10].
Our focus was implementing Data Standards in a study using CDISC. CDISC is an independent organization established in 1997 to improve research by introducing methods and practices of data interpretation and standardization [11].
Database build and testing
Database designing and CRF designing show a positive correlation. The eCRF enables the entry of data into the relational database. Once a CDMS is selected for a study, Data Requirement Specifications (DRS) are created by the Data Manager. These specifications serve as a backbone for the database creation.
Data Validations
Data Validations play a major role in ensuring the data is comprehensive and of high quality to meet the study’s objectives and comply with the regulatory standards. A Data Validation Specification (DVS) is created based on the Data Requirement Specification. It contains validation checks to ensure that all possible data fields capture data per protocol and discrepancies if any are highlighted as a ‘query’. Edit/Automatic checks are embedded in the clinical database to detect the discrepancies in the entered data and thus ensuring data validity. In e-CRF based trial, data validation process as known as query management process is a consistent ongoing process to identify discrepancies in the data [1,8]
Conduct phase
Once the study database is developed with all validations in place and first patient first visit takes place the conduct phase begins. The data collected from patient’s visit per the visit schedule are entered in the database.
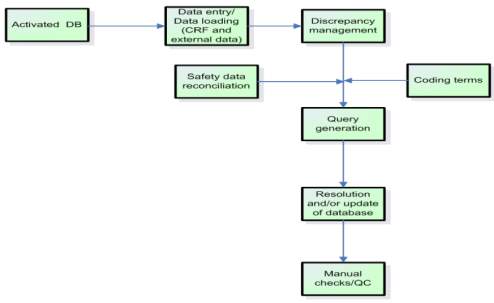
Figure 4: An overview on clinical data management conduct activities (Clinical Data Management- An introduction, QA Data, www.quadata.co.za)
The main components of this phase are:
1. Discrepancy Management
It is a process of cleaning subject data in the CDMS by programmed edit checks or manual checks.Data manager raises queries for discrepant data and the sites correct the data/ provide a clarification. Once an issue is resolved, query is closed.
CRF tracking, Medical Coding, Serious Adverse Event (SAE) Reconciliation form other components of conduct phase.
Closeout phase
This phase involves resolving all pending issues in the database and locking the database. The locked data is then extracted and sent to the statisticians for analysis.
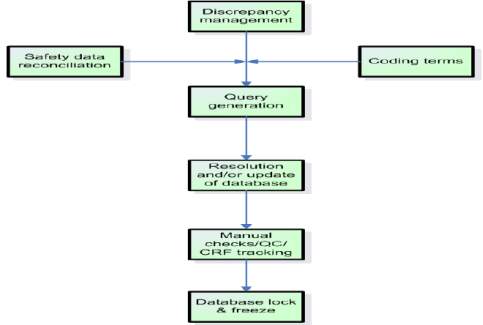
Figure 5: An overview on clinical data management closeout activities (Clinical Data Management- An introduction, QA Data, www.quadata.co.za)
Details outlined above clearly demonstrate the heavy involvement of CDM in providing high quality clinical data. It is obvious that if one would want to ensure richness in data quality, proactive planning from study set up is essential.
With ICH-GCP E6(R2) coming into picture, the industry has now turned its attention to improved planning. Regulatory bodies are spearheading efforts to ensure study quality. As a result, sponsors are dedicating effort and time to developing a Quality Management System throughout all stages of clinical trial process [12].
While quality system exists in the industry, there is academic conceptual framework for clinical data quality management that aims to address quality and monitor and improve performance in sponsor driven academic clinical development environment.
With current project we aimed to develop a robust QMS for sponsor driven academic studies in compliance with ICH-GCP E6(R2) [10,12].
1.4 Research Aim and Objectives
To develop two integral components of Clinical Data Management Quality Systems in compliance with ICH-GCP E6 (R2) addendum for an academic set up:
1.4.1 Implement and test international data standards (CDISC SDTM) during clinical study set up
We aimed at implementing global data standards using CDISC Terminology with an aim to downstream the standards in all the sponsor based academic studies in the organisation. The approach was to incorporate these standards during the CRF annotation phase of start-up. The objective was to make it easier for regulatory bodies to review data across studies and to ensure all studies conform to the same high standard. Having a standardized structure would also make it easier to check the integrity of the data from a data management perspective.
1.4.2 Develop a robust Quality Control Plan to ensure consistent high clinical data quality collection during all three Clinical Data Management phases – Set up, Conduct and Closeout.
To ensure that project is executed successfully according to SDTM standards, heavy manual efforts by data management team and the database programming team are required. This is bound to lead to data integrity issues. To ensure that these issues are minimized, it becomes extremely important to have a contingency plan in place. In clinical data management such a contingency plan is called as Quality Control Plan (QCP). This plan is a document that cumulatively specifies quality standards, specifications of activities according to all phases of clinical trials (Start-up, Conduct/ Maintenance, Closeout).
At a highest level, quality goals should be aligned along with overall strategic plans of the organization which keep client and sponsors in business as you deliver data with highest quality.
At lowest level, quality plan resembles an actionable plan which is in parallel with organizations demand and desirable outcome.
2. MATERIALS AND METHODS
Two separate studies were used as implementation models for each component of the research.
- To Implement and test international data standards (CDISC SDTM) during clinical study set up- Risk Based Screening for the Evaluation of Atrial Fibrillation Trial (R-BEAT) trial was used as an implementation model
- To Develop a robust Quality Control Plan to ensure consistent high clinical data quality collection during all three Clinical Data Management phases – Set up, Conduct and Closeout- Clarifying Optimal Sodium Intake Project 1 (COSIP-1) was used as a testing model
As the project was designed to develop two integral components of QMS in compliance with ICH-GCP E6 (R2) addendum for an academic set up, it was essential to select studies that were sponsored by the organisation in which the research was carried out, and the set-up had to be academic in nature.
The project was carried out at the Health Research Board (HRB) Clinical Research Facility Galway (CRFG), National University of Ireland (NUI) Galway.
Both R-BEAT and COSIP-1 were non-regulated studies that belonged to HRB CRFG.
Implementation and testing of international data standards (CDISC SDTM) during clinical study set up
Implementation of data standards can only be done in set up phase, it was essential to use a study in set-up phase to test the implemented standards. This was another reason why R-BEAT was selected for testing.
R-BEAT
Study Title: Risk Based Screening for the Evaluation of Atrial Fibrillation Trial (R-BEAT)
Protocol Version: 1.1
Co-Principal Investigators: Professor Martin O’Donnell & Dr. Ruairi Waters, HRB Clinical Research Facility Galway (CRFG), National University of Ireland, Galway
Study Design:
A randomized controlled cross-over multi-centered clinical trial in General Practice, comparing; a) opportunistic pulse screening + immediate ELR device (R-test) for 2 weeks; or, b) opportunistic pulse screening + delayed ELR device (R-test) for 2 weeks, in patients with CHA2DS2-VASc score of 3 or greater, and without a contraindication to oral anticoagulation.
Recruitment was undertaken under informed consent, following eligibility assessment.
The Galway University Hospitals Ethics Committee reviewed the study protocols and related information sheets, to ensure your rights as a participant are protected.
Data was protected in compliance with General Data Protection Regulation (GDRP).
Research Question:
Primary research question
Will extended cardiac rhythm monitoring (with ELR for 2-weeks), compared to standard care, in patients pre-identified to be at high-risk of atrial fibrillation (defined by CHA2DS2-VASC score >2) increases the detection of new atrial fibrillation, that is efficient, acceptable to patients, and cost-effective.
Secondary research question
Will self-screening with regular pulse checks identify patients with high-risk of atrial fibrillation, in an embedded sub-study (SWAT).
Study Objectives:
- To determine whether a risk-based screening programme for occult paroxysmal atrial fibrillation, involving extended cardiac monitoring in adults with CHA2DS2-VASc score of 3 or greater, increases the detection of new atrial fibrillation/flutter.
- To determine whether a risk-based screening programme for occult paroxysmal atrial fibrillation, involving extended cardiac monitoring in adults with CHA2DS2-VASc score of 3 or greater, is cost-effective.
- To determine the sensitivity, specificity, positive predictive value and negative predictive values of self-monitoring of pulse in adults for detection of atrial fibrillation.
- To determine the cost, cost effectiveness, and budget impact of a risk-based screening programme for occult paroxysmal atrial fibrillation, relative to a control of usual care in general practice.
Eligibility Criteria:
Inclusion criteria
- Signed a study specific Informed Consent Form
- At least 55 years of age.
- All adults attending one of the participating General Practices in the R-BEAT Trial.Attended at least one GP appointment within the last year.
- CHA2DS2-VASC Score >2
Exclusion criteria
- Contraindication to oral anticoagulant therapy
- History of intracerebral haemorrhage
- Prior intolerance or refusal of oral anticoagulant therapy
- Gastrointestinal haemorrhage of unexplained or unmodifiable aetiology (i.e. risk of haemorrhage has not been reduced)
- Other major bleed
- Unsuitable for anticoagulant therapy, in opinion of attending general practitioner
- Cardiac monitoring for >48 hours in the last year
- Patient has an Implantable Loop Recorder
- Unsuitable for cardiac monitoring, in opinion of attending general practitioner
- Allergies to plasters or adhesives
Once an approved study protocol was received from the project co-ordinator, DRS creation was initiated per following workflow.
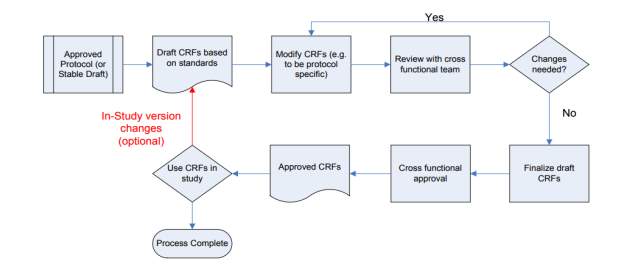
Figure5: CRF creation process flow
Critical data items for the study were determined. Using this critical data field information and the study visit schedule from protocol, the process of annotation implementation and DRS creation was done as per the CDISC SDTM standards. All critical data field from this study based on its Primary and Secondary outcome were taken into consideration while framing annotation. The aim was to use this DRS for CRF creation and ultimately set standard annotation rules across the organization to increase the quality of annotations.
| CRF form | Field |
| CHA2DS2-VASc Score | All fields |
| Medical history | All fields |
| Medical events | All fields |
| Adverse events | All fields |
| Study withdrawal | All fields |
| R-TEST RESULTS | All fields |
Critical data fields taken into consideration: –
Table 1. Critical data fields from R-BEAT protocol considered for DRS creation
Defining Standards and conventions
As CRF pages were annotated to maintain consistency with each variable and domain, there were many variables (annotations) which were unique to the study and were not found in the CDISC repository. During such instances, Clinical Data Acquisition Standards (CDASH) library of specific domain was used in one of the following ways:
- Direct: – CDM variable were copied directly from the CDISC repository to a domain variable without any changes
- Rename: – Variable name and label was changed
- Splitting: – CDM variable (annotation) were divided into two or more SDTM variables
- Combining: – To form a single SDTM variable directly two or more CDM variables were combined
Each domain had unique two-character code which represented domain
| Domain | SDTM annotation |
| Adverse events | AE |
| Demographics | DM |
| Medical History | MH |
| Physical Examination | PE |
Table 2. Examples of SDTM two-code character and the corresponding domain
Each observation corresponding to its domain was prefixed by its domain ID. For Example: – MHTERM. MH defined domain it belonged to and TERM defined observation it collected for that domain. Using this methodology each variable and its functionality was defined.
Annotation for repeating variables were created once and were used with different domains. For example: STRT (Start Date). Start date variable holds annotation of STRT and this was used across study to maintain consistency. Start date was a very common and most repeating variable in the database as every visit would have a start date. To map one variable to one single visit, STRT was used as suffix for all domains and the domain ID was used as a prefix.
As a result, while designing a DRS, for demographic the start date annotation was DMSTRT (DM-prefix + STRT suffix) and for Adverse Event start date annotation was AESTRT (AE-prefix + STRT suffix).
The designed DRS contained different sections and some generic rules and limitation were followed while creating this document
- Module/Section Title– Has a maximum of 15 characters with no spaces, a mix of upper and lower case. Example Medical History.
- Variable Label– Each datapoint for that was expected to be collected
- Variable Alias– Annotation per CDISC SDTM. Maximum 40 characters were allowed with no spaces in between.
- Field Type– Captured Single/ Multiple response (example if only one option was expected to be collected in a CRF a single response would be programmed and if multiple options were expected to be collected in a CRF then field type would be multiple response).
- Field Length– Every database has some restrictions in terms of characters that can be captured for that variable. This section depicted format and conventions used to capture date and time.
- Category Values– As data goes for regulatory submission it is be structured and consistent, instead of free text to capture responses numerical conventions were used to identify and limit response capture for variable. These values provided a unique nomination to each data field response when more than one response could be collected- ‘Yes’ and ‘No’.
- Mandatory/Optional– Defined if a datapoint is mandate or not.
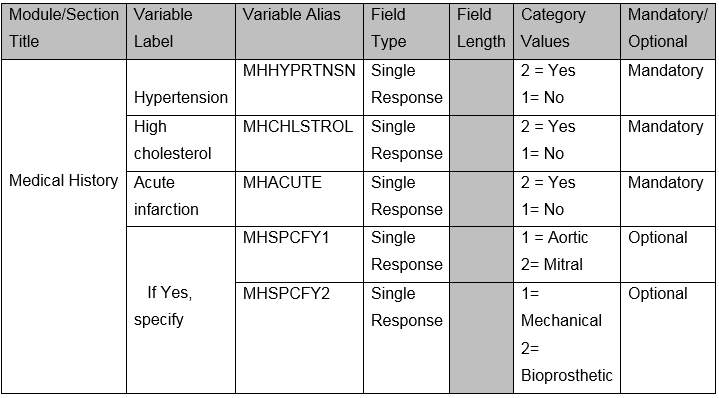 Table 3. Example of DRS for Medical History
Table 3. Example of DRS for Medical History
Above figure shows how the final DRS would look like. The DRS created served as a data repository which guided the database developer for mapping variables to CRF creation.
Development and Testing of a robust Quality Control Plan on COSIP-1 study
A Quality Control Plan to analyse data integrity was implemented for each phase of clinical data management phase i.e. Setup / Maintenance and Closeout phase. Checkpoints were designed to analyse data variance at each phase of clinical trial. A working QCP was designed listing each activity and errors categories.
All the data management activities were planned and listed in the QCP. Checkpoints for all activities were shortlisted depending on the impact on quality. Ideology behind implementing QC plan was to monitor and execute activity with minimal error. Once a task is performed by a data manager same activity was performed by an independent reviewer to analyse variance. Each activity was categorized according to their phase and criticality.
The plan was divided in three sections i.e. Setup phase / Maintenance / Closeout phase which illustrated set parameters that would meet the sponsor’s clean data requirement. All activities were analysed based on their criticality (Major and Minor) which served as measure of variance.
Study Title: Clarifying Optimal Sodium Intake Project 1 (COSIP-1)
Protocol Version: 6.0
Co-Principal Investigators: Professor Martin O’Donnell & Professor Andrew Smyth, HRB Clinical Research Facility Galway (CRFG), National University of Ireland, Galway
Study Design:
A Phase IIb randomised, 2-group, parallel, open-label, controlled trial conducted at a single-centre with an allocation ratio of 1:1.
Research Question:
In adult participants, is an educational intervention to reduce sodium intake targeting low intake levels, compared to targeting moderate intake (control), associated with changes in a panel of cardiovascular biomarkers, over 2 years follow-up?
Study Objectives:
1. To determine whether a low-sodium diet (
2. To determine the effect of low sodium diet (compared to moderate sodium intake) on 24-hour ambulatory blood pressure by completing 24-hour blood pressure monitoring at randomisation (T0) and two years (T8)
Eligibility Criteria:
Inclusion Criteria:
(i) Age 40 years or older
(ii) Systolic blood pressure
(iii) No change in anti-hypertensive or diuretic medications (including dose) for 3 months before screening visit
(iv) Self-reported willingness to modify dietary intake over sustained period, and adhere with directed recommendations over 2 years
(v) Signed written informed consent
Exclusion Criteria:
(i) Known chronic kidney disease (CKD) or most recent eGFR ≤60ml/min/1.73m2 a. Participants who are ineligible for COSIP based on their eGFR will be approached about entering the ongoing Sodium Intake in Chronic Kidney Disease (STICK) trial.
(ii) Any of the following renal conditions:
- Glomerular disease due to post-infectious glomerulonephritis, IgA nephropathy, thin basement membrane disease, Henoch-Schonlein purpura, proliferative glomerulonephritis, membranous nephropathy (including lupus nephritis), rapidly progressive glomerulonephritis, minimal change disease, or focal segmental glomerulosclerosis
- Prior or planned renal transplant
- Prior, current or planned renal dialysis
(iii) Previous cardiovascular disease:
- Myocardial infarction
- Previous percutaneous coronary intervention (PCI), percutaneous transluminal coronary angioplasty (PTCA), or coronary artery bypass grafting (CABG)
- Stroke (previous transient ischaemic attack [TIA] is not an exclusion criterion)
(iv) Medical diagnosis known to be associated with abnormal renal sodium excretion, including the following:
- Bartter syndrome
- SIADH
- Diabetes insipidus
(v) Serum sodium
vi) Severe heart failure defined as NYHA Class III/IV OR left ventricular ejection fraction (LVEF) ≤30%
(vii) High-dose loop or thiazide diuretic therapy, exceeding a total daily dose of frusemide 80mg, bumetanide 2mg, hydrochlorothiazide 50mg, bendroflumethiazide 2.5mg, indapamide 2.5mg, metolazone 2.5mg or the use of both a loop and thiazide diuretic
(viii) Unable to follow educational advice of the research team
(ix) Prescribed high-salt diet, low-salt diet or sodium bicarbonate
(x) Symptomatic postural hypotension or receiving treatment for postural hypotension
(xi) Current or recent use (within one month) of immunosuppressive medications including tacrolimus, cyclosporine, azathioprine or mycophenolate mofetil
(xii) Pregnancy or lactation
(xiii) Unable to comply with 24-hour urinary collections, or medical condition making collection of 24-hour urinary collection difficult (e.g. severe urinary incontinence)
(xiv) Participant unlikely to comply with study procedures or follow-up visits due to severe comorbid illness or other factor (e.g. inability to travel for follow-up visits, drug or alcohol misuse) in the opinion of the research team
(xv) Cognitive impairment defined as a known diagnosis of dementia or inability to provide informed consent due to cognitive impairment in the opinion of the investigator
(xvi) Body Mass Index (BMI) 40 kg/m2
(xvii) Participating in another clinical trial or previous allocation in this study
Since the DRS was created in the first part of research, the functioning of QCP was demonstrated on the DRS creation process. Although the QCP was designed for all three phases of Clinical Data Management. Each component of the plan was designed in agreement with the data management and development team at HRB Clinical Research Facility-Galway.
For a certain project, DRS can be designed by any individual involved in the study. It was inevitable that the data and document standards may differ from one person creating the document to the other. This would ultimately hamper data quality and documentation quality at the same time. It thus was essential to assure that same set of standards and instructions were followed to have consistency across the projects.
The designed QCP contained following rows:
1. Activity– This column consisted activities under review-DRS creation in this case
2. Frequency– Frequency of activity being performed, in this case since DRS creation was performed only once, the Final DRS document was considered
3. Sample size– DRS is considered one of the critical data management documents during study set-up and thus 100% QC was performed
4. Check points– Based on DRS limitation criteria and annotation standards checkpoints were designed in agreement with the data management team at HRB Clinical Research Facility Galway.
5. Error Rate– Each error was classified according to its impact on database development. For example- An error of ‘variable label’ missing could hamper database development timeline or variable label not meeting 40 characters limitation could stop database programming. These errors were categorized as major errors and issues which needed attention but would not impact database designing process were categorised as minor. To maintain consistency in error reporting, the acceptable error rate for DRS creation process was determined as ≤ 4 Major error or ≤ 8 Minor error. If the error rate went above this limit for a project, the DRS creation process was expected to be reconsidered.
6. Classification– Depending on the errors rates identified, they were documented as major or minor.
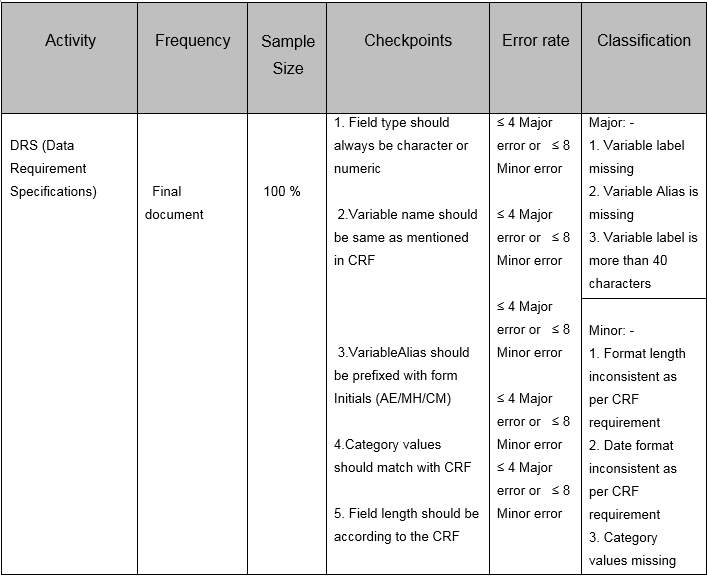
Table 4. Example of QCP for DRS creation process
3. RESULTS and DISCUSSION
Part A: SDTM naming convention implemented Data Requirement Specification
The SDTM naming convention/ data annotations were defined in the DRS for all the data fields for R-BEAT. However, it was difficult to demonstrate the entire DRS document in the thesis, as a result only 6 critical fields (Modules/Section)- ‘CHA2DS2VASScore’, ‘Medical Events’, ‘Medical History’, ‘Adverse Event’, ‘Study Withdrawal’ and ‘Test Result’have been shown below. These variables were designed for idatafax CDMS as idatafax was the chosen CDMS for R-BEAT. The applied standards followed idatafax standards along with SDTM. Each component of DRS was categorized as Module/Section Title, Variable Label, Variable Alias, Field Type, Field Length, Category Values and Mandatory/Optional.
Below is the output of the DRS for each critical field described individually:
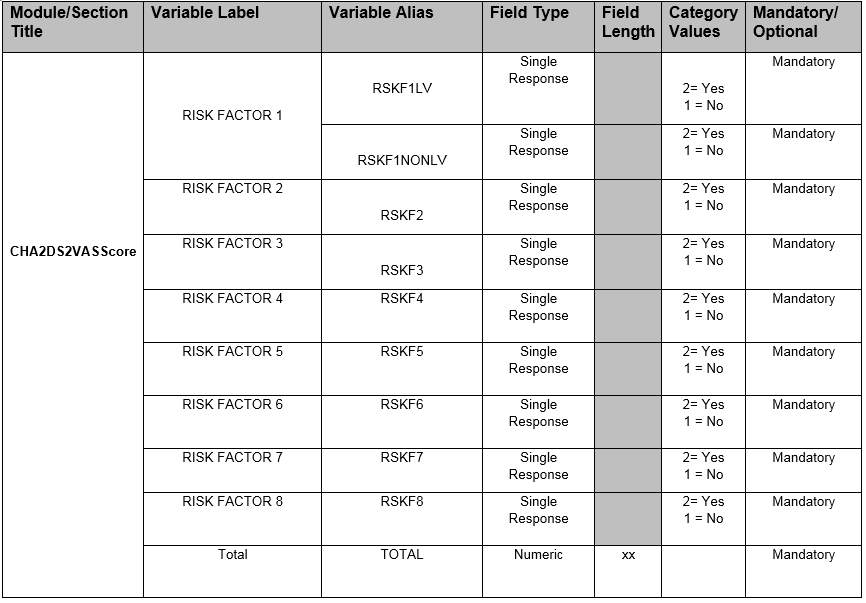
Table 5 – CHA2DS2VASScore SDTM implemented Data requirement specification
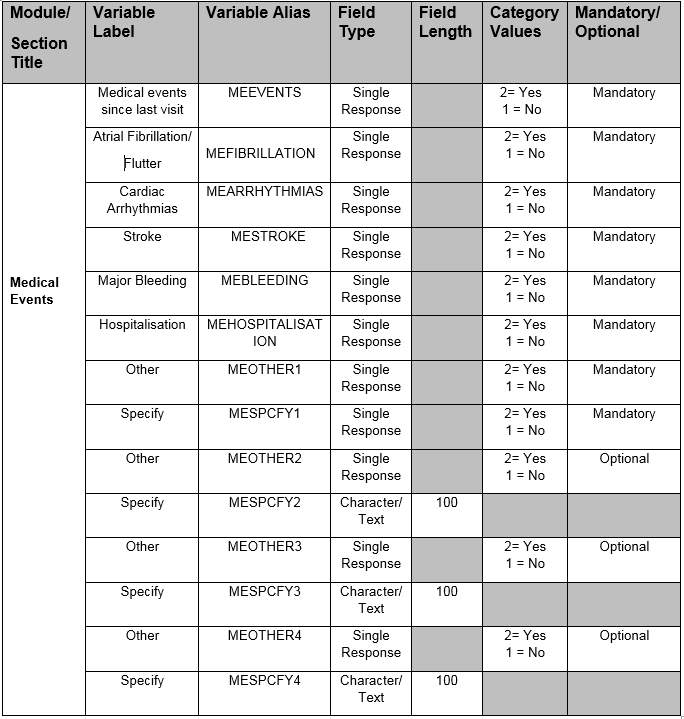
Table 6 – Medical Events SDTM implemented Data requirement specification
| Module/
Section Title |
Variable Label | Variable Alias | Field Type | Field Length | Category Values | Mandatory/ Optional |
|
Medical History
|
Hypertension | MHHYPRTNSN | Single Response | 2= Yes 1 = No | Mandatory | |
| High cholesterol | MHCHLSTROL | Single Response | 2= Yes 1 = No | Mandatory | ||
| Angina /PCI | MHANGNPCI | Single Response | 2= Yes 1 = No | Mandatory | ||
| Myocardial Infarction | MHMYCINFRACTN | Single Response | 2= Yes 1 = No | Mandatory | ||
| Ischaemic retinal stroke | MHSTROKE | Single Response | 2= Yes 1 = No | Mandatory | ||
| Transient attack | MHTRNST | Single Response | 2= Yes 1 = No | Mandatory | ||
| Venous | MHVENOUS | Single Response | 2= Yes 1 = No | Mandatory | ||
| Acute infarction | MHACUTE | Single Response | 2= Yes 1 = No | Mandatory | ||
| Heart Valve replacement | MHHEART | Single Response | 2= Yes 1 = No | Mandatory | ||
| If Yes, specify | MHSPCFY1 | Single Response | 1 = Aortic 2= Mitral | Mandatory | ||
| MHSPCFY2 | Single Response | 1= Mechanical 2= Bioprosthetic | Mandatory | |||
| Syncope | MHSYNCOPE | Single Response | 2= Yes 1 = No | Mandatory | ||
| Hypothyroidism | MHHYPOTHYDYSM | Single Response | 2= Yes 1 = No | Mandatory | ||
| Hyperthyroidism | MHHYPRTHDYSM | Single Response | 2= Yes 1 = No | Mandatory | ||
| Diabetes mellitus | MHDIABETES | Single Response | 2= Yes 1 = No | Mandatory | ||
| Fall in last 12 months | MHFALL12MNTH | Single Response | 2= Yes 1 = No | Mandatory | ||
| Heart palpitations | MHHRTPALP | Single Response | 2= Yes 1 = No | Mandatory | ||
| If Yes, participant felt palpitation | MHPRTCPTPALP | Single Response | 2= Yes 1 = No | Mandatory |
| Module/
Section Title |
Variable Label | Variable Alias | Field Type | Field Length | Category Values | Mandatory/ Optional |
| Medical History
|
If Yes, palpitation occurrence | MHPALPOCCR | Single Response | 1 = Daily 2= Weekly 3=Monthly 4= Rarely | Mandatory | |
| Prior echo available | MHECHOPR | Single Response | 2= Yes
1 = No |
Mandatory | ||
| If yes, ejection fraction | MHEJCFRACTION | Numeric | xx | Mandatory | ||
| Dilated Left Atrium | MHDLTLFTATRM | Single Response | 2= Yes
1 = No |
Mandatory | ||
| Left atrial size | MHLFTATRLSIZE | Numeric | xx.x | Mandatory |
Table 7 – Medical History SDTM implemented Data requirement specification
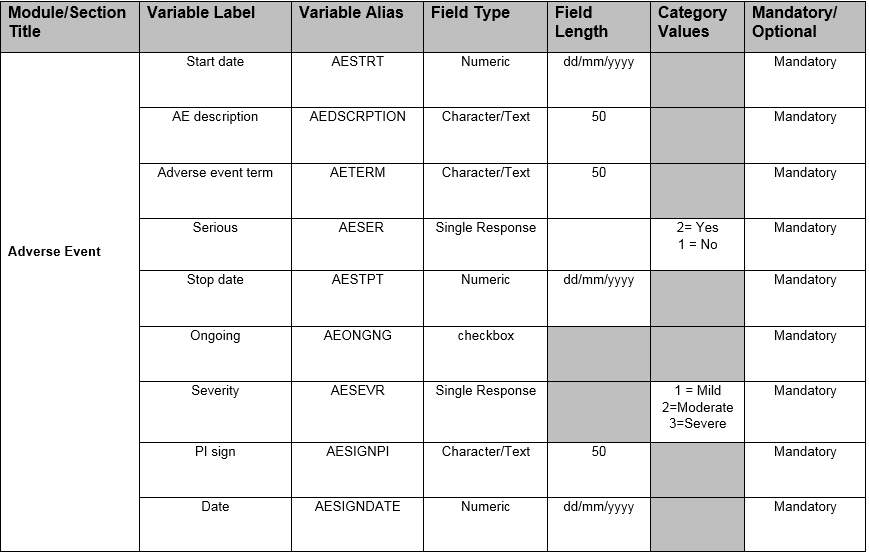
Table 8 – Adverse Events SDTM implemented Data requirement specification
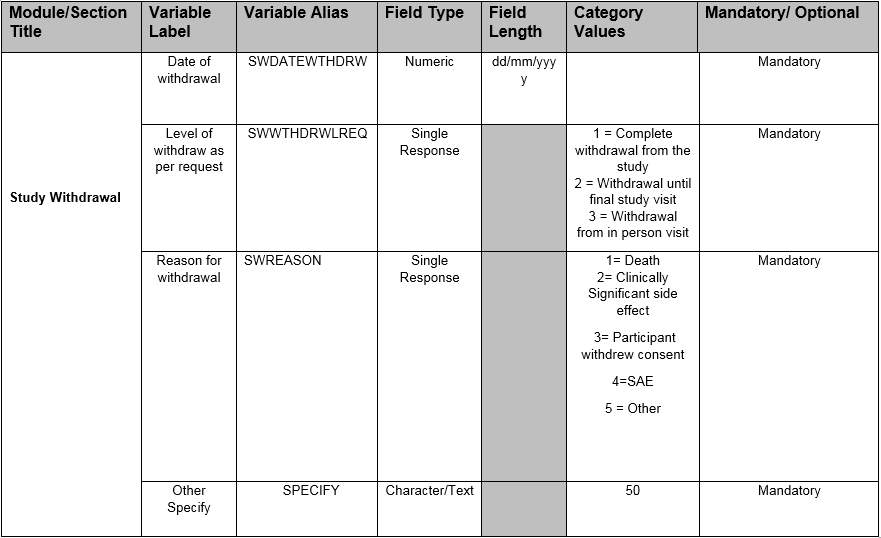
Table 9 –Study Withdrawal SDTM implemented Data requirement specification
| Module/Section Title | Variable Label | Variable Alias | Field Type | Field Length | Category Values | Mandatory/ Optional |
| Test
Results |
Transmission Start | RTRNSMSSON | Date | dd/mm/yyyy | Mandatory | |
| Transmission Time | RTRNSMSSONTIME | Time | hh:mm | Mandatory | ||
| Recorder Number | RTESTRCRD | Numeric | xxxxxxxxxxx | Mandatory | ||
| Monitor Duration | RTESTMNTRDUR | Time | HHH:MM:SS | Mandatory | ||
| Analyzed Duration | RTESTANADUR | Time | HHH:MM:SS | Mandatory | ||
| Heart rate Min | RTESTHRTRTMIN | Numeric | xxx | Mandatory | ||
| Heart rate Mean | RTESTHRTRTMEAN | Numeric | xxx | Mandatory | ||
| Heart rate Max | RTESTHRTRTMAX | Numeric | xxx | Mandatory | ||
| Atrial Fibrillation/Flutter | RTESTATRLFIBLLTN | Single Response | 2= Yes 1 = No | Mandatory |
| Module/Section Title | Variable Label | Variable Alias | Field Type | Field Length | Category Values | Mandatory/ Optional |
|
Test Results
|
If yes, select | RTESTCRITERIA | Single Response | 1= Atrial Fibrillation 2= Atrial flutter 3=Uncertain | Mandatory | |
| If yes. Episodes | RTESTEPISODES
|
Numeric
|
xxxx
|
Mandatory | ||
| Longest duration | RTESTDURATION
|
Time | mm:ss
|
Mandatory | ||
| Burden | RTESTBURDEN | Single Response | 1= 2 MIN-24HR 4= >24 HR | Mandatory | ||
| IF >2 MIN-24 HR; length | RTESTLNGTH
|
Time | hh:mm:ss
|
Mandatory | ||
| IF >2 MIN-24 HR estimate
|
RTESTESTIMATE
|
Checkbox | Mandatory | |||
| Were any detected | RTESTDETCTION | Single Response | 1= MULTIPLE PAC 2=SVT 3= VENTRICULAR TACHYCARDIA 4= VENTRICULAR FIBRILATION 5 =PAUSE | Mandatory | ||
| LONGEST PAUSE DETECTED | RTESTLONGPAUSE
|
Numeric
|
xxx.xx
|
Mandatory |
Table 10 –Test Results SDTM implemented Data requirement specification
Benefits of developing a sponsor defined SDTM interpretation guide
When a sponsor develops an interpretation guide for SDTM, one will notice several “quick wins”. First one being having an SDTM compliant study with documented interpretation and clarified rules. One can develop and streamline programming methods for many of the SDTM domains through template or macro programs. Especially for an academic set up, as the documentation improves, and experience grows, the efficiency will as well. Besides the from programming benefits, the SDTM domains produced from these guidelines will be more compliant and readier for regulatory submission.
Even more importantly, if SDTM implementation standards and sponsor rules are followed, it will academically be simple to develop a repository warehouse that is also SDTM compliant. From this repository, periodical safety updates and exploratory analysis can be performed. One can also rapidly perform any ad-hoc evaluation. If the repository is consistent and with meaningful interpretation, one can also answer regulatory questions that involve multiple studies [9].
Part B: Implementation of Quality Control Plan on non-standardized DRS
| Module | Variable Label | Error description | Category |
| Medication | Variables name are not annotated, not as per generic annotation standards | Major | |
| Category values to capture field response missing (Example: – Mild /Moderate / severe) | Major | ||
| Field type should predict characteristic of field (Example: – Character / Numeric/ Single response / Checkbox) | Major | ||
| Only Mandatory or Optional can be recorded for fields. | Major | ||
| Module name with XXXX is not as per standards, it should represent variable for CRF | Major | ||
| Collect if conditions missing as per DRS format. (IF MEDICATION OTHER = OTHER, SPECIFY NOT UPDATED) | Major | ||
| Variable name is not as per generic annotation standards | Major | ||
| Intervention | Field XX | Alphanumeric characters not acceptable in variable | Major |
| Variable name is not as per generic annotation standards | Major | ||
| Field type – Date format should always be in DDMMYYYY format | Major | ||
| Field Length should only be in numeric representing only allowable length to capture data | Minor | ||
| Blood results | Full CRF Question captures variable options which is not as per Datafax standards | Major | |
| Date format not mentioned in the document | Major |
Figure 11: – Data requirement specification errors post implementing Quality Control Plan
QC tool was designed and implemented to the DRS for COSIP-1. It was not possible to demonstrate all the modules QC’d in the thesis, as a result we only showed three major modules for demonstration (i.e., medication, intervention and blood results). There were 12 major and one minor findings observed for these three modules. This clearly illustrated the requirement of streamlining database related document creation process.
When nature of these findings was carefully analysed, it was seen that the process of DRS creation was not standard throughout the organisation. The focus of implementing quality checkpoint was to identify typical errors subsequently regulate and create framework that can be implemented for all studies making the database development process consistent throughout the organisation.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Information Systems"
Information Systems relates to systems that allow people and businesses to handle and use data in a multitude of ways. Information Systems can assist you in processing and filtering data, and can be used in many different environments.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: