Pharmacotherapy for Heart Failure
Info: 10719 words (43 pages) Dissertation
Published: 9th Dec 2019
Tagged: MedicinePharmacology
Pharmacotherapy for heart failure
Epidemiology of Heart Failure
Heart failure continues to be a rapidly growing global epidemic with an estimated prevalence of more than 37.7 million individuals worldwide. According to American Heart Association (AHA), heart failure (HF) has an estimated prevalence of 5.7 million in the USA among adults ≥ 20 years of age. This figure is expected to rise by as much as 46% by the year 2030 resulting in more than 8 million patients with heart failure. Annually 870,000 new HF cases are recorded and the lifetime risk of developing HF in people aged 40 years is 1 in 5 in both males and females. The annual cost of caring for HF was estimated to be $30.7 billion in 2012, with as much as 68% attributable to direct medical costs. These costs have been projected to increase by nearly 127% to $69.7 billion by the year 2030.
Globally, heart failure is common throughout Sub-Saharan Africa where 44% of newly diagnosed cardiovascular disease patients have HF as compared to 10% who have IHD. In these areas, the average age at involvement of HF patients is much less as compared to United States and Europe. The prevalence rates of HF range from 1.26% to 6.7% across Asia.
Based on Framingham heart study, a joint project of the National Heart, Lung and Blood Institute and Boston University, McKee et al. in 1971 proposed a set of standardized criteria for HF for use in research studies. The study data highlighted the importance of advancing age and raised blood pressure as a precursor of heart failure and demonstrated poor long term outcomes in these patients with nearly half of affected individuals dying within 5 years of diagnosis.
The incidence and prevalence data for heart failure have remained unreliable in most developing countries because of the lack of robust surveillance systems that track incidence, prevalence, risk factors and outcomes of heart failure. Despite this inadequacy, there is a general consensus on the escalating burden of HF in these regions on account of ‘epidemiological transition’ from infectious and acute diseases to chronic illnesses, aging populations and increasing prevalence of risk factors like hypertension, coronary artery disease, diabetes and obesity. In India, the estimated prevalence of heart failure due to coronary artery disease (CAD), hypertension, obesity, diabetes mellitus and rheumatic heart disease (RHD) ranges from 1.3-4.6 million with the annual incidence ranging from 0.5 to 1.8 million (Huffman and Prabhakaran, 2010).
Management of heart failure
The goals of pharmacotherapy for heart failure are to alleviate symptoms, slow or reverse deterioration in myocardial function, improve patient’s functional status and quality of life, minimize hospitalizations, and to reduce disease mortality. Medical care for heart failure includes management of contributing factors and associated conditions, lifestyle modification, pharmacologic, and invasive interventions to limit and reverse the cardinal manifestations. Lifestyle modification primarily focuses on smoking cessation, restriction of alcohol, salt and fluid intake, physical activity as deemed appropriate and weight management. Reduction of symptoms can often be achieved with diuretics, beta blockers, angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), angiotensin receptor-neprilysin inhibitors (ARNI), aldosterone receptor antagonists, hydralazine and nitrate, and digoxin. Evidence for survival benefit is currently available for certain beta blockers (bisoprolol, carvedilol, and metoprolol), ACE inhibitors, ARBs, ARNI, combined hydralazine-nitrate therapy and aldosterone receptor antagonists. While cardiac glycosides are useful in chronic systolic heart failure, other positive inotropic agents are indicated primarily in acute systolic failure. The use of latter, as demonstrated in large clinical trials, is associated with reduced survival and/or no benefit in chronic heart failure and is discouraged.
Pathophysiological mechanisms of heart failure and role of pharmacotherapeutic interventions
The understanding of pathophysiological mechanisms of heart failure has advanced significantly over the past several decades from hemodynamic model to neurohormonal paradigm. In response to a reduced cardiac output, several neurohormonal compensatory responses are activated to maintain adequate cardiac output, including stimulation of the sympathetic nervous system activity and the renin–angiotensin aldosterone pathway, resulting in increased vascular tone, salt and water retention as well as ventricular hypertrophy and remodeling. These compensatory mechanisms that support cardiovascular function, however, paradoxically contribute to vicious spiral progression of heart failure. A schematic illustration of major pathophysiologic mechanisms and sites of action of drugs is given below:
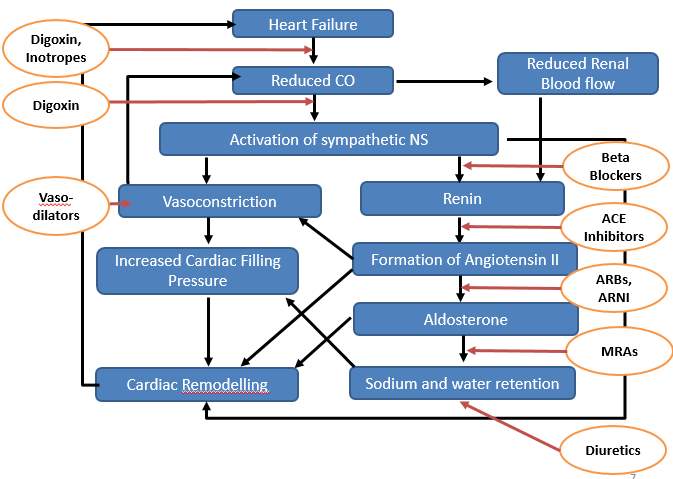
Drugs for the management of heart failure can be classified into those useful for chronic heart failure and acute decompensated heart failure (Table 1.)
| Pharmacological treatment of Heart Failure | |
| Chronic Systolic Heart Failure | Acute Decompensated Heart Failure |
| Diuretics | Diuretics |
| Aldosterone Receptor Antagonists | Vasodilators |
| ACE inhibitors | Beta-adrenergic and Dopaminergic Agonists |
| Angiotensin Receptor Blockers | Phosphodiesterase inhibitors |
| Angiotensin Receptor-Neprilysin Inhibitors (ARNI) | Natriuretic Peptide (Nesiritide) |
| Beta blockers | |
| Digoxin | |
| Vasodilators | |
Pharmacotherapy for chronic systolic heart failure
Angiotensin-Converting Enzyme (ACE) Inhibitors
The first Angiotensin-Converting Enzyme (ACE) Inhibitor to be marketed in 1977, captopril is an early example of drug design based on knowledge of target molecular structure. Various small peptide molecules were known to be weak inhibitors of ACE. Based on this knowledge, captopril was designed to combine their steric properties with a non peptide structure enabling oral administration. While captopril has a short half life and needs to be administered twice or thrice daily, the newer agents have longer duration of action allowing once daily administration. Findings from several large clinical trials support their use in the management of CHF of any severity, including cases with asymptomatic left ventricular dysfunction. To reduce mortality and morbidity, ACE inhibitors are recommended in all patients with heart failure with reduced ejection fraction with current or prior symptoms, unless contraindicated.
Mechanism of action:
Angiotensin converting enzyme inhibitors act by blocking the conversion of angiotensin I to angiotensin II leading to blunting of its vasoconstriction and sodium retaining effects (Fig. 1).The decrease in angiotensin II and aldosterone reduces many of the deleterious effects of these neurohormones and leads to potential benefits. ACE inhibitors reduce vascular resistance, improve tissue perfusion and reduce afterload. By decreasing aldosterone production and the direct stimulatory effect of angiotensin II on reabsorption of sodium and bicarbonate in the early part of the proximal convoluted tubule, they attenuate salt and water retention and reduce preload. Lowering of tissue angiotensin levels also reduces sympathetic over activity through reduction of angiotensin’s presynaptic effects on norepinephrine release. ACE inhibitors also increase bradykinin levels indirectly leading to enhanced prostaglandin production, contributing to its effects on reducing peripheral vascular resistance. Accumulated angiotensin I is also directed towards alternate metabolic pathways leading to increased production of Ang (1-7), a vasodilatory peptide. Importantly these drugs reduce myocardial fibrosis, myocyte apoptosis and long-term remodelling of the heart and vessels, effects that are deemed responsible for the clinically observed reduction in mortality and morbidity in heart failure patients.
Figure 1: Mechanism of action of ACE inhibitors and Angiotensin Receptor Blockers
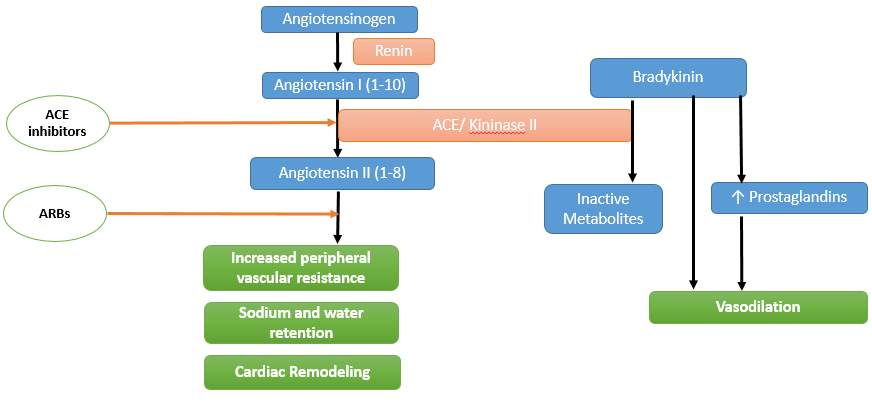
Clinical Use:
Since ACE inhibitors have been shown to reduce the risk of death as well as hospital admissions in patients with heart failure, it is widely recommended that ACE inhibitors be prescribed to all patients with reduced ejection fraction unless they are contraindicated or not tolerated by the individual. Their benefits have been observed in all subsets of patients – with mild, moderate, or severe symptoms of CHF and in individuals with or without coronary artery disease. To avoid causing symptomatic hypotension due to elevated renin activity in these patients, ACE inhibitors should be initiated at low doses followed by gradual increments in dose once the lower doses are well tolerated. Patients should undergo assessment of their renal function and serum potassium within 1 to 2 weeks of initiation of therapy and periodically thereafter especially in cases with pre-existing hypotension, hyponatremia, diabetes mellitus, azotemia or those taking potassium supplements. An important clinical consideration is determining the appropriate dose of ACE inhibitor in CHF patients. The clinical trials that established the clinical utility of these agents uptitrated the drug doses to pre-determined target levels rather than according to patient’s therapeutic response. Although efforts should be made to reach the target dose shown to be effective in major clinical trials with these agents, this may at times be limited by development of hypotension and/or decline in renal function. In these cases where target doses cannot be used, intermediate/ lower doses should be used with the knowledge that there are likely only small differences in mortality outcomes between low and high doses. Abrupt withdrawal of treatment with these agents should be avoided since it can lead to deterioration of clinical status.
The initial and maximum doses of these agents are mentioned in the table 1 below (1). Individual pharmacokinetic parameters of these agents are depicted in table 2.
Table 1: Dosing guide for commonly used ACE inhibitors
| ACE Inhibitors | Initial Dose | Maximum Dose |
| Captopril | 6.25 mg thrice daily | 50 mg thrice daily |
| Enalapril | 2.5 mg twice daily | 10 to 20 mg twice daily |
| Fosinopril | 5 to 10 mg once daily | 40 mg once daily |
| Lisinopril | 2.5 to 5 mg once daily | 20 to 40 mg once daily |
| Perindopril | 2 mg once daily | 8 to 16 mg once daily |
| Quinapril | 5 mg twice daily | 20 mg twice daily |
| Ramipril | 1.25 to 2.5 mg once daily | 10 mg once daily |
| Trandolapril | 1 mg once daily | 4 mg once daily |
Adverse effects:
Adverse effects directly attributable to ACE inhibition are common to all drugs of this class. This includes hypotension, especially after first dose of ACE inhibitor can be seen in heart failure patients particularly if they have been treated with loop diuretics leading to strong activation of their renin angiotensin system. Elevated bradykinin levels from ACE inhibition are associated with dry cough, the commonest adverse effect. In such cases, substitution of ACE inhibitor with an angiotensin receptor blocker is often helpful. A more serious consequence of kinin accumulation is angioedema, a potential life threatening side effect which may occur at any time during the course of therapy and warrants immediate and permanent cessation of all ACE inhibitors. Patients with bilateral renal artery stenosis develop renal failure when treated with these drugs because in them, despite low afferent arteriolar pressure, glomerular filtration is maintained by angiotensin II by selectively increasing efferent arteriolar tone. A mild increase in serum potassium is common with ACE inhibitors. Such cases may be managed by instituting low potassium diet but may require drug dosage adjustment. ACE inhibitors are avoided in patients who are pregnant or who plan to become pregnant.
Table 2: Pharmacokinetic Parameters of Commonly Used ACE inhibitors
| ACE Inhibitors | Prodrug/ Active Metabolite | Time to peak plasma concentration (hours) | Half-life (hours) | Bioavailability (%) | Metabolism | Excretion | Adjustment in renal failure | Adjustment in hepatic failure |
| Captopril | No/NA | 1-1.5 | 2 | 65 | Liver (50%) | Renal (95%) | Yes | No |
| Enalapril | Yes/Enalaprilat | 4* | 11* | 60 | Liver (70%) | Renal (61%)
Hepatic (33%) |
Yes | Yes/No # |
| Fosinopril | Yes/ Fosinoprilat | 3* | 11-13* | 30-36 | Liver | Renal (44%)
Hepatic (46%) |
No | No |
| Lisinopril | No/NA | 7 | 12 | 25 | Liver (7%) | Renal (29%)
Hepatic (69%) |
Yes | No |
| Perindopril | Yes/ Perindoprilat | 3-4* | 25* | 75 | Liver (90%) | Renal (75%)
Hepatic (25%) |
Yes | No |
| Quinapril | Yes/ Quinaprilat | 2* | 3* | 50 | Liver (Extensive) | Renal (55%)
Hepatic (33%) |
Yes | Yes/No # |
| Ramipril | Yes/ Ramiprilat | 2-4* | 13-17* | 60 | Liver (Extensive) | Renal (40-60%)
Hepatic (40%) |
Yes | No |
| Trandolapril | Yes/ Trandolaprilat | 4-6* | 16-24* | 10 | Liver (Extensive) | Renal (33%)
Hepatic (66%) |
Yes | Yes |
*For active metabolite
# Depending on severity of liver dysfunction. Expect reduced efficacy of drug with increased liver dysfunction
References:
- APC/DTC Briefing Document. January 2008. NHS
- Angiotensin-converting enzyme inhibitor (ACE inhibitor) class review. October 2001. McGill University Health Centre
- Brunton LL, Chabner B, Knollmann BC. Goodman and Gilman’s Pharmacological Basis of Therapeutics. McGraw Hill Education. 12th Edition. 2011.
- Katzung BG, Trevor AJ. Basic & clinical pharmacology. McGraw-Hill Medical. 13th Edition. 2016.
Angiotensin II Receptor Blockers
Angiotensin II Receptor Blockers (ARBs) were developed based on the rationale that angiotensin II production continues even in the presence of ACE inhibition driven by non-ACE-dependent pathways e.g., chymase, cathepsin, and kallikrein allowing increase in the circulating levels of angiotensin II, the so called “ACE-escape” phenomenon. Losartan, candesartan, valsartan, irbesartan, telmisartan, olmisartan and eprosartan are the currently available non-peptide, orally active AT1 receptor ARBs.
Mechanism of action:
Based on their differential binding affinity for antagonists and cellular actions, two distinct subtypes of angiotensin II receptors – AT1 and AT2 have been identified. While most of well known actions of angiotensin II viz. vasoconstriction, SNS activation, aldosterone release, and sodium and water retention are mediated through AT1 receptors. However effects of AT2 receptor activationhave now been recognized and include vasodilation, development of fetal tissues, cell differentiation, apoptosis, and inhibition of cell proliferation (Fig below). The ARBs by blocking the angiotensin II receptor subtype, AT1, prevent the detrimental effects of angiotensin II, regardless of its origin. They also allow unopposed activation of AT2 receptors which causes effects nearly opposite to those of AT1 receptor stimulation. Additionally, blocking the receptors rather than ACE would produce all of the benefits of ACE inhibitors while minimizing the probability of developing bradykinin mediated side effects including cough and angioedema. However, some of the beneficial effects of ACE inhibitors are attributable to accumulation of kinins rather than inhibition of angiotensin II generation, like kinin induced vasodilation (Refer to figure in ACE inhibitors section).
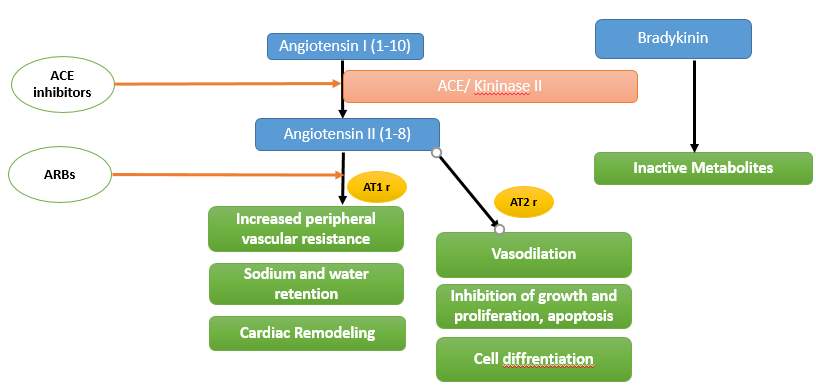
Clinical Use:
In systolic HF patients, ACE inhibitors remain the first choice for inhibition of renin angiotensin system since they have a more robust evidence base, but angiotensin II receptor blockers are now considered to be reasonable alternatives. In several placebo controlled studies, long term treatment with ARBs has been shown to reduce hospitalizations as well as mortality in CHF patients. Owing to their favourable adverse effect profile, ARBs are an excellent alternative in patients with CHF who are ACE inhibitor intolerant, the most common indication being cough. Angioedema is a potential life threatening side effect which may occur at any time during the course of ACE inhibitor therapy and requires immediate and permanent cessation of all ACE inhibitors. Although ARBs may be considered as alternative options for such patients, caution is always advised in these cases as angioedema, although rarely, has been reported with the use of AT1 receptor antagonists as well. Guidelines recommend against routine combination therapy with ACEi, ARB and MRA. However, cautious use of ACEi and ARB combination is advised in a select group of symptomatic HFrEF patients who are MRA intolerant and who are also using beta blockers.
Treatment with ARBs is initiated at low doses with similar considerations as in case of ACE inhibitors including monitoring of blood pressure, renal function and serum potassium levels within 1 to 2 weeks after initiation and also after changes in dose. In general, uptitration is carried out by doubling the doses. Close surveillance is advised in patients with low systolic blood pressure (<80 mm Hg), low serum sodium, diabetes and impaired kidney function.
The usual initial and maximum doses of ARBs are mentioned in the table below (1).
| Angiotensin Receptor Blocker | Initial Dose | Maximum Dose |
| Candesartan | 4 to 8 mg once daily | 32 mg once daily |
| Losartan | 25 to 50 mg once daily | 50 to 150 mg once daily |
| Valsartan | 20 to 40 mg twice daily | 160 mg twice daily |
Adverse effects:
Angiotensin II receptor blockers are generally well tolerated and have lower incidence of angioedema and cough as compared to ACE inhibitors. In patients with renal artery stenosis, ARBs may cause oliguria, progressive azotemia or acute renal failure. ARBs may cause elevated potassium levels in patients with renal impairment and in those taking potassium supplements or potassium sparing diuretics. They have teratogenic potential and should be avoided in pregnancy and in patients who plan to become pregnant.
Beta adrenergic receptor antagonists (Beta Blockers)
For several years, CHF treatment involved drugs with sympathomimetic properties. This was based on the belief that heart failure is fundamentally a disorder of reduced stroke volume and cardiac output. Long-term use of sympathomimetics was expected to improve clinical outcomes based on relief obtained from short term use of dopamine and dobutamine. Under this model, the use of beta adrenergic receptor antagonists was believed to be counterintuitive; however, the reverse appears to be the case in the clinical settings. Chronic sympathomimetic use is associated with increased mortality rates and there is considerable evidence from several placebo controlled randomized clinical trials that beta-blockers reduce morbidity and mortality in patients with heart failure. Long term use of these agents can improve HF symptoms, clinical status as well as patient’s overall sense of well being. Benefits have been observed in patients with or without CAD, with or without diabetes mellitus and also in those already taking ACE inhibitors. Three beta blockers have been shown to significantly reduce risk of death compared with placebo: carvedilol, sustained release metoprolol , and bisoprolol.
Mechanism of action:
The mechanisms by which beta receptor antagonists improve outcomes in CHF patients have not been fully elucidated. The suggested mechanisms include i) prevention of myocardial ischemia, ii) attenuation of cytotoxic and signaling effects of high concentrations of catecholamines, iii) inhibition of renin release,iv) antiarrhythmic effects,v) reduction or reversal of ventricular remodelling through inhibition of mitogenic activity of catecholamines and vi) induction of positive LV remodelling by attenuating oxidative stress in the myocardium. While bisoprolol and metoprolol selectively block beta-1–receptors; carvedilol blocks alpha-1, beta-1, and beta-2–receptors.
Clinical Use:
It is recommended that beta blockers be prescribed to all patients with stable heart failure with reduced ejection fraction unless they are contraindicated or not tolerated by the individual. Initiation of a beta-blockers at usual doses in patients with heart failure patients can potentially lead to symptomatic worsening or acute decompensation due to their negative inotropic effects. Therefore beta-blockers should be initiated at very low doses followed by gradual increments in dose. Patients should be carefully monitored for changes in vital signs and symptoms during this increment period. Planned upward titration in the dose of a beta blocker should be delayed until any untoward effects observed with lower doses have disappeared in these patients. Even when symptoms do not improve with beta blockers, long-term treatment needs to be maintained to minimize the risk of major clinical events. The initial and maximum doses of these agents are mentioned in the table below (1). Efforts should be made to reach the target dose shown to be effective in major clinical trials with these agents. Although several beta blockers are available, their clinical benefit in HF patients should not be assumed to be a class effect. Buccindol, for example failed to show consistent results across different patient populations while nebivolol did not reduce mortality alone when evaluated in a randomized trial enrolling elderly HF patients with reduced and preserved LVEF.
| Beta Blocker | Initial Dose | Maximum Dose |
| Bisoprolol | 1.25 mg once daily | 10 mg once daily |
| Carvedilol | 3.125 mg twice daily | 50 mg twice daily |
| Carvedilol CR | 10 mg once daily | 80 mg once daily |
| Metoprolol succinate extended release (CR/XL) | 12.5–25 mg once daily | 200 mg once daily |
Adverse effects:
Major adverse effects encountered in heart failure patients initiated on beta blocker therapy include fluid retention and worsening of heart failure, fatigue, bradycardia or heart block and hypotension. Patients presenting with fluid retention or worsening symptoms generally respond favourably to intensification of conventional therapy and once treated they remain excellent candidates for long term treatment with beta blockers. Bradycardia produced by beta blockers is generally asymptomatic and requires no intervention; however, if it is accompanied by dizziness or light headedness or if second- or third-degree heart block occurs, reduction in the dose of beta blocker is warranted. Risk of hypotension may be minimized by taking beta blocker and ACE inhibitor at different times during the day. Hypotensive symptoms may also respond to a decrease in the dose of diuretics in volume depleted patients. The symptom of fatigue is multifactorial and may be related to other causes such as sleep apnoea, excessive diuresis, or depression.
Mineralocorticoid receptor antagonists (MRA)
Reduced cardiac output in CHF patients via activation of renin angiotensin system may lead to circulating aldosterone levels that are nearly 20 times above normal. The pathophysiological effects of raised aldosterone levels in these patients are diverse and include sodium and water retention, increased loss of K+ and Mg2+, myocardial and vascular fibrosis, reduced uptake of norepinephrine in the myocardium, baroreceptor dysfunction and alterations in sodium channel expression in cardiac myocytes leading to increased excitability and contractility. Angiotensin II is not the sole stimulus for aldosterone secretion and its level have been shown to return to pretreatment values upon chronic treatment with ACE inhibitors, a phenomenon termed as ‘aldosterone escape’. These observations provided the rationale for combining aldosterone receptor antagonists with ACE inhibitors and other standard therapy and led to their evaluation in several major randomized clinical trials with the aim of investigating their effects on mortality and HF hospitalizations in patients with CHF with reduced ejection fractions (Fig.).
| RCT | Year | Drug | Target dosage | No. of patients | LVEF | NYHA class | Primary end-point(CV deaths and HF hospitalizations) |
| RALES | 1999 | Spironolactone | 25-50 mg QD | 1,663 | ≤35% | III-IV | 38.1% vs 50.5% |
| EPHESUS | 2003 | Eplerenone | 25-50 mg QD | 6,642 | ≤40% | II-IV | 26.6% vs 30% |
| EMPHASIS -HF | 2011 | Eplerenone | 25-50 mg QD | 2,737 | ≤30%, ≤ 35% if QRS > 130 ms | II | 18.3% vs 25.9% |
Mechanism of action:
Mineralocorticoid receptor antagonists also known as aldosterone receptor antagonists compete with aldosterone to bind at its mineralocorticoid receptor and were originally developed as potassium sparing diuretics. However later studies demonstrated additional benefits based on disruption of deleterious effects of aldosterone mentioned above. Although the exact mechanisms by which aldosterone receptor antagonists improve heart failure morbidity and mortality remain elusive, neurohormonal benefits contribute largely since the doses recommended in these cases have minimal diuretic effects. Spironolactone is a nonselective aldosterone receptor antagonist marketed since the 1960s and bears structural similarity to progesterone resulting in sex steroid receptor cross reactivity. This is responsible for anti-androgen and anti-progesterone adverse effects seen in treated individuals. Canrenone, marketed as principle active metabolite of spironolactone, is devoid of significant first pass metabolism and has a substantially longer t ½ as compared to the parent molecule (16.5 hours vs 1.4 hours). It has also been reported to have a lower incidence of antiandrogenic adverse reactions despite being a non-selective MRA. Eplerenone, a spironolactone derivative, in contrast is a selective aldosterone receptor antagonist with lower affinity for the progesterone and androgen receptors and consequently is devoid of related adverse effects. Eplerenone is primarily metabolized in the liver by cytochrome P450 (CYP3A4 isoenzyme) and is therefore subject to interactions with CYP3A4 inhibitors as well as inducers. It has a plasma half-life of 4-6 hours, and steady state levels are usually achieved 48 hours after treatment initiation.
Clinical Use:
Aldosterone receptor antagonists are currently recommended in NYHA class II–IV CHF patients with left ventricular ejection fraction ≤35%. To be considered for therapy with these agents, patients with NYHA class II heart failure should have a previous history of cardiovascular related hospital admission or increased plasma natriuretic peptide levels. Serum creatinine should be ≤ 2.5 mg/dL in males and ≤ 2.0 mg/dL in females, and serum potassium should be less than 5.0 mEq/L. Careful monitoring of patient’s kidney function and serum potassium is warranted in these patients at both initiation of therapy as well as during maintenance. Aldosterone receptor antagonists are also recommended in patients of acute myocardial infarction with left ventricular ejection fraction ≤40% and symptomatic heart failure or a diagnosis of diabetes mellitus.
The recommended doses of these agents are mentioned in the table below (1).
| MRA | eGFR (mL/min/
1.73 m2) |
Initial Dose | Maintenance Dose |
| Spironolactone | ≥ 50 | 12.5 – 25.0 mg once daily | 25 mg once or twice daily |
| 30-49 | 12.5 mg once daily or every other day | 12.5 – 25.0 mg once daily | |
| Eplerenone | ≥ 50 | 25 mg once daily | 50 mg once daily |
| 30-49 | 25 mg once every other day | 25 mg once daily |
Adverse effects:
The major adverse effect associated with MRA therapy is hyperkalemia due to their potassium sparing effects. Consequently they are contraindicated in hyperkalemic patients or those at high risk of developing hyperkalemia either due to disease or due to concomitant medications. Risk of serious hyperkalemias in HF patients may be minimized by routine serum potassium and kidney function monitoring. Development of serum potassium levels > 5.5 mEq/L is considered to be indication of lowering drug dose or stoppage of MRA therapy unless other causes are present. Development of worsening renal function should be managed with evaluation of entire regimen and if need be interruption of MRA therapy. Patients should also discontinue therapy during episodes of diarrhoea, dehydration or when loop diuretics are interrupted. Additionally, spironolactone may cause gynecomastia, breast tenderness, erectile dysfunction, reduced libido, hirsutism, deepening of voice and menstrual irregularities. It may also cause diarrhoea, gastritis, peptic ulceration, ataxia, confusion, skin rashes and blood dyscrasias rarely. As mentioned above, CYP3A4 inhibitors or inducers should be avoided in patients receiving eplerenone.
Diuretics
The compensatory mechanisms ensuing in heart failure patients lead to excessive sodium and water retention, often resulting in pulmonary as well as systemic congestion. Diuretic therapy, in addition to salt restriction, remains crucial in managing congestive symptoms in such patients. They have no direct effect on cardiac contractility and their major therapeutic benefit involves reduction in venous pressure and ventricular preload. However, there is no evidence of a mortality benefit or altered disease progression in CHF patients receiving either loop or thiazide diuretics. Heart failure patients often operate on the plateau portion of Frank-Starling curve, reduced preload improves congestive symptoms but has little effect on the stroke volume or cardiac output. Since overdiuresis can potentially reduce cardiac output, renal perfusion and cause dehydration, diuretics are best avoided in patients with asymptomatic left ventricular dysfunction. Minimal doses required to maintain euvolemia should be employed in patients symptomatic from hypervolemia.
Mechanism of action:
Loop diuretics
Among the currently available agents, furosemide, torsemide, and bumetanide are the principal diuretics used in the treatment of heart failure. Ethacrynic acid, on account of its ototoxicity is generally reserved for patients with sulfonamide allergies or those intolerant to other available options. These drugs act by inhibiting Na+-K+-2Cl– symporter (NKCC2) in the thick ascending limb of the loop of Henle, resulting in increased sodium, potassium, and water excretion. These agents are highly bound to plasma proteins and are not significantly filtered at the glomerulus. They are actively transported by the organic acid transport pathway to reach the tubular lumen. They also induce a prostaglandin mediated increase in renal blood flow, which contributes to their natriuretic effect. The latter effect may be blocked by co-administered non-steroidal anti-inflammatory drugs resulting in diminished diuretic efficacy.
Thiazide diuretics
Thiazide diuretics like hydrochlorothiazide inhibit sodium and chloride reabsorption via the Na+Cl– cotransporter (NCC) in the distal convoluted tubule. As compared to loop diuretics, they are relatively weak diuretics and are seldom prescribed alone in CHF patients. They may be used in combination with loop diuretics in cases refractory to loop diuretics alone. These agents may be preferred in patients with mild fluid retention and hypertensive heart disease because of their more persistent antihypertensive actions.
Potassium sparing diuretics
Potassium sparing diuretics inhibit Na+ influx through ion channels in the luminal membrane of renal epithelial cells (amiloride, triamterene) or are antagonists at mineralocorticoid receptors (spironolactone, and eplerenone). Together these agents are weak diuretics, but are useful in achieving volume reduction with limited potassium wasting. The beneficial effects of aldosterone receptor blockers on mortality reduction in CHF patients are discussed separately.
Clinical Use:
Furosemide has a variable oral absorption ranging from 10% to 100%. Maximal response to loop diuretics is generally reduced in CHF patients. This is believed to result in part from impaired gut absorption, as may occur in patients with CHF-induced gastrointestinal edema, and/or increased tubular reabsorption of sodium, possibly ascribed to increased Na-K-2Cl transporter activity. Thus, it is recommended that more frequent dosing be employed once the ceiling dose is attained for additional benefit rather than giving progressive higher dosages. As compared to furosemide, bumetanide and torsemide have demonstrated superiority in reducing symptoms such as dyspnea and fatigue as well as greater weight loss. Significant reduction in the hospital readmission rates and all-cause mortality were also observed. These results may be attributed to the greater bioavailability of torsemide and bumetanide — both in excess of 80%. Torsemide, additionally has a longer half-life than both furosemide and bumetanide. The appropriate chronic dose is one that maintains the patient at a stable dry weight without dyspnoeic symptoms.
The usual daily dose of furosemide ranges from 20-160 mg/day. The usual initial dose is 20-80 mg given as single dose. If needed, the same dose may be repeated 6-8 hours later or dose may be raised by 20-40 mg. The individually determined single dose should then be prescribed once or twice daily. When doses in excess of 80 mg/day are prescribed for prolonged duration, careful laboratory monitoring of serum electrolytes, kidney function tests as well as clinical observations are recommended. For torsemide, usual initial oral dose is 10-20 mg once daily. The dose should be titrated upward by approximately doubling until the desired response is obtained in HF patients. Maximum total daily dose is limited to 200 mg. The usual total daily dosage of bumetanide is 0.5-2 mg and in most patients it is given as a single dose. In case of an inadequate response, a second or third dose may be given at 4-5 hour intervals up to a maximum daily dose of 10 mg.
Adverse effects:
Adverse effects of loop diuretics that are related to their efficacy include hyponatremia and/or ECF volume depletion and consequent hypotension, reduced GFR, circulatory collapse, thromboembolic episodes, and hepatic encephalopathy in patients with liver disease. In addition, increased urinary K+ and H+ excretion causes hypochloremic alkalosis. Resultant hypokalemia may induce arrhythmias and lead to increased mortality, particularly in patients taking cardiac glycosides. Hypomagnesemia and hypocalcemia can also occur in these patients on account of increased urinary losses.
Loop diuretics can cause ototoxicity clinically presenting as tinnitus, hearing impairment, deafness, vertigo and fullness in the ears. Hearing impairment and deafness are often reversible. Ototoxicity is more common with rapid intravenous administration as compared to oral administration. These agents may also cause hyperuricemia, hyperglycemia and can adversely affect lipid profile of patients by increasing low-density lipoprotein and triglyceride levels while reducing high-density lipoprotein levels. They are also associated with interstitial nephritis and skin reactions.
Drug interactions may occur when loop diuretics are co-administered with other ototoxic drugs viz. aminoglycosides, carboplatin, paclitaxel; anticoagulants – increased anticoagulant effects, lithium – increased risk of lithium toxicity, NSAIDs and probenecid – reduced diuretic response.
Angiotensin Receptor-Neprilysin Inhibitors (ARNI)
Natriuretic peptides (NP) are hormones responsible for maintaining cardiorenal homoeostasis due to their natriuretic, diuretic, vasodilatation, and other properties. Among the four type of these peptides, atrial natriuretic peptide (ANP) is released by the atria in response to myocardial stretching caused by increased intravascular volume and brain natriuretic peptide (BNP) is released by the ventricles in response to increased ventricular volume and pressures. Both ANP and BNP have similar physiological actions i.e. diuresis, natriuresis, vasodilatation and reduced catecholamine and renin secretion. The third peptide, C-type natriuretic peptide (CNP) is released by the endothelium of the heart, lungs, kidneys, and vessels but is not found in circulation in significant concentrations. CNP has less diuretic capabilities but is a potent vasodilator and prevents vascular cell growth. Finally, D-type NP is also related to CHF, but not much is known about its physiology and significance. Natriuretic peptides are degraded by the neutral endopeptidase neprilysin, which also degrades bradykinin, substance P, adrenomedullin, glucagon, vasoactive intestinal peptide, and also contributes to breakdown of angiotensin II. Although NP levels are elevated in CHF patients due to fluid overload, these concentrations are ineffective at providing clinical benefit and symptom relief. One method of further increasing NP levels satisfactorily is to reduce their breakdown through inhibition of neprilysin. To avoid counterproductive effects of excess angiotensin II, neprilysin must be combined with an inhibitor of the renin–angiotensin–aldosterone system. Omapatrilat, a dual inhibitor of neprilysin as well as ACE, failed to gain regulatory approval due to severe cases of angioedema, likely attributable to markedly elevated levels of bradykinin. The novel combination drug valsartan/sacubitril (LCZ696) is a first-in-class angiotensin receptor-neprilysin inhibitor that has recently gained FDA approval for the treatment of heart failure under the brand name Entresto.
Mechanism of action:
LCZ696 is a crystalline complex comprised of the neprilysin inhibitor sacubitril and the angiotensin receptor blocker valsartan in their anionic forms, sodium cations, and water molecules in the molar ratio of 1:1:3:2.5 (Figure). Following oral ingestion, LCZ696 dissociates into sacubitril, a prodrug which is enzymatically cleaved to the active neprilysin inhitor, LBQ657 and valsartan. As compared to omapatrilat, the combination drug is designed to have a decreased risk for angioedema because it inhibits only one of the enzymes responsible for bradykinin degradation.
Figure (Ref: JCHF. 2014;2(6):663-670. )
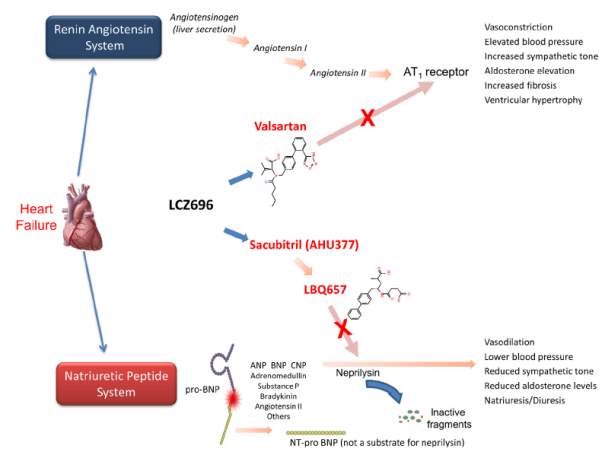
The approval of this novel compound was based on a large double-blind, randomized controlled trial comparing sacubitril-valsartan (200 mg bid) and enalapril (10 mg bid) in patients with chronic systolic heart failure designated as the PARADIGM-HF trial. This study included more than 8000 patients who were randomized to receive either of the active treatments in addition to guideline-directed medical therapy comprising of beta blockers and mineralocorticoid receptor antagonists. The primary composite outcome (cardiovascular death or first hospitalization for heart failure) was reported in 21.8% of the patients in the sacubitril-valsartan treatment group vs 26.5% of patients in the enalapril treatment group. The study found significant reductions in the primary outcome (20%), cardiovascular death (20%), and all-cause mortality (16%) with novel treatment vs. enalapril.
Clinical Use
The recommended starting dose of sacubitril/valsartan is 49/51 mg twice daily. Thereafter the dose is doubled after 2-4 weeks to target dose of 97/103 twice daily based on patient’s tolerance. In patients not previously taking ACE inhibitor or ARB or those taking low doses of these agents, patients with severe renal impairment and moderate hepatic impairment, a starting dose of 24/26 mg twice daily is recommended. Subsequently, the dose is doubled every 2-4 weeks to the target maintenance dose of 97/103 mg twice daily, as tolerated by the patient. The drug is usually prescribed concomitantly with other HF therapies in substitution of an ACE inhibitor or other ARB.
Adverse effects
The main adverse effects noted in more than 5% clinical trial participants were hypotension, hyperkalemia, cough, dizziness, and renal failure. The combination can cause fetal toxicity and should be discontinued as soon as pregnancy is detected. Angioedema was experienced by 0.5% patients in the sacubitril-valsartan group and 0.2% patients in the enalapril group.
Ivabradine
The heart rate lowering agent, Ivabradine, underwent a fast track evaluation and received FDA approval for the treatment of chronic HF in April 2015. It has been authorized for use by European Medicines Agency since 2005 and included in European Society of Cardiology guidelines since 2012. Ivabradine is a first-in-class agent with a novel mechanism of action – by selective inhibition of the pacemaker If “funny” channel, which is responsible for the automaticity of the sinoatrial node. Ivabradine is recommended for patients with symptomatic stable HF with left ventricular ejection fraction less than/ equal to 35 percent, in sinus rhythm with a resting heart rate greater than/ equal to 70 beats per minute, and who are either are on a beta blocker at maximally tolerated doses or have an intolerance/ contraindication to beta blocker use. In such patients, concomitant treatment should include ACE inhibitor (or ARB), and a mineralocorticoid receptor blocker.
Mechanism of Action
Based on observational cohorts and randomized trials, reduction of heart rate is a potential therapeutic target in patients with HF with reduced ejection fraction since an increased heart rate has been associated with morbidity, mortality and adverse cardiovascular outcomes. An elevated heart rate reflects, activation of the sympathetic activity and withdrawal of activity of parasympathetic nervous system, which are components of the neuro-humoral response to reduced cardiac output.
Ivabradine’s molecular target of action is the hyperpolarization-activated cyclic nucleotide-gated (HCN) channel. These channels create the diastolic depolarization or “funny” pacemaker current (If) of the SA node, named so because it is characteristically activated by the membrane hyperpolarization following systolic depolarization. Ivabradine, in a use dependent manner, directly blocks HCN channels thus inhibiting the cardiac pacemaker current in the SA node, prolonging diastolic depolarization and ultimately reducing the heart rate in healthy as well as diseased hearts, both at rest and with exertion. The cardiac actions of ivabradine are selective for the SA node, and the drug is devoid of effects on blood pressure, myocardial contractility, ventricular repolarization, intra-atrial, atrioventricular or intraventricular conduction time. Reduction in heart rate results in prolonged diastolic perfusion time, improved coronary blood flow and enhanced exercise capacity and in contrast to beta blockers, ivabradine increases stroke volume. It also improves heart rate variability and exerts anti-remodelling effect as demonstrated in both preclinical and clinical studies.
Safety and efficacy of ivabradine has been established by two major randomized trials (CARNIVA HF and SHIFT) and an open label multicenter study (INTENSIFY). The pivotal Systolic Heart failure treatment with the If inhibitor Ivabradine Trial (SHIFT) was a multi-centric, randomized, double-blinded, placebo controlled study. The trial results revealed favorable outcomes of ivabradine therapy with 18% reduction in the primary composite end point of cardiovascular death and hospital admission due to worsening HF.
Clinical Use
Ivabradine causes a dose dependent reduction in HR with greatest reduction occurring in patients with highest HR at baseline. Its tablets are available in two strengths – 5 mg and 7.5 mg. The recommended starting dose is 5 mg twice daily administered with meals. In patients with conduction defects or in whom reduction in HR could result in hemodynamic compromise, the starting dose is 2.5 mg twice a day. Those patients who are also receiving a beta blocker, should continue at the same dose.
Following initiation of treatment, the patient is assessed after 2 weeks and dose is adjusted to achieve a resting HR between 50 and 60 bpm. Dose modifications are made as per the table below:
| Patient heart rate (bpm) | Dose modification | |
| 1 | More than 60 | Dose increased by 2.5 mg given twice daily up to a maximal dose of 7.5 mg BD |
| 2 | Between 50 – 60 | Dose is maintained |
| 3 | Less than 50 or signs and symptoms of bradycardia | Dose is decrease by 2.5 mg given twice daily; if current dose is 2.5 mg twice daily, therapy is discontinued. |
Adverse effects
In the SHIFT trial, the most common reported adverse events were bradycardia, hypertension, atrial fibrillation and phosphenes. Phosphenes are off target visual effects of ivabradine characterized by transient sensations of increased brightness in a restricted area of visual field. They are believed to result from inhibition of retinal Ih currents, that have properties similar to those of cardiac If currents. They are usually reported with 2 months of treatment initiation, are reversible and not associated with treatment discontinuation. In the post-approval phase, adverse events reported with ivabradine include syncope, hypotension, torsade de pointes, ventricular tachycardia/fibrillation, angioedema, erythema/ rash, itching, urticaria, vertigo, visual impairment and diplopia.
Ivabradine is predominantly metabolized in liver and intestine by CYP 3A4 and therefore concomitant administration of strong CYP inhibitors viz. antifungal azoles, macrolide antibiotics, HIV-protease inhibitors and nefazodone is to be avoided. The drug is also contraindicated in patients with acute decompensated HF, BP less than 90/50, severe hepatic dysfunction, pacemaker dependence, and in patients with sick sinus syndrome, SA block or 3rd degree heart block (unless a functioning demand pacemaker is in place) due to increased risk for bradycardia.
Nitrates and Hydralazine
Although several vasodilators have been developed for the management of CHF patients, only ACE inhibitors, ARBs and hydralazine-isosorbide dinitrate combination have demonstrated survival benefits. A combination of isosorbide dinitrate and hydralazine is recommended in patients with symptomatic heart failure with reduced ejection fraction in addition to standard therapy when ACE inhibitor (or ARB) cannot be given because of intolerance or contraindication viz. angioedema, renal insufficiency or hyperkalemia.
Mechanism of Action
The complimentary hemodynamic actions of nitrates and hydralazine provide the rationale for this combination therapy. Nitrates serving as nitric oxide donors activate soluble guanylyl cyclase in vascular smooth muscle resulting in increased cGMP, venodilation and consequently reduced preload. In addition they reduce pulmonary and systemic vascular resistance albeit at higher doses, dilate epicardial coronary arteries — enhancing blood flow and improving ventricular function. These pharmacodynamics effects result in symptomatic benefit and improved exercise tolerance. However, when used alone their clinical utility is rapidly limited by development of tolerance. But when used in combination with hydralazine, there is a synergistic increase in clinical effectiveness and sustained improvement of CHF hemodynamics. For hydralazine, the precise mode of action is not yet fully characterized. It is believed to be a directly acting arterial vasodilator with little actions on venous capacitance vessels. In CHF, hydralazine reduces systemic as well as pulmonary vascular resistance resulting in reduced afterload and improve stroke volume and cardiac output. Interestingly, it also possesses direct positive inotropic effects on cardiac muscles independent of its vascular actions. Since hydralazine increases renal blood flow to a greater extent when compared to other vasodilators with the exception of ACE inhibitors, it is preferred for the management of CHF patients with renal impairment who cannot tolerate ACE inhibitors. Hydralazine when used in combination, also limits nitrate tolerance through an antioxidant effect that enhances NO levels by attenuating superoxide formation.
The strength of evidence of benefit with hydralazine and isosorbide dinitrate (ISDN) combination is stronger for African American patients as compared to other ethnicities. A relative small RCT that exclusively enrolled men demonstrated that hydralazine and ISDN, each administered four times daily reduced mortality compared to placebo in patients also receiving diuretics and digoxin. The trial was conducted before the advent of ACE inhibitors and beta blockers as standard treatment for HF. A subsequent trial conducted in self-identified patients of African descent found that supplementation of a fixed dose combination of hydralazine and isosorbide dinitrate to conventional therapy (ACE inhibitor/ ARB, Beta blocker and MRA) reduced mortality and hospitalizations in HFrEF patients.
Clinical Use
Treatment with a fixed dose combination (FDC) containing 20 mg of ISDN and 37.5 mg hydralazine can be initiated with one tablet three times daily. If well tolerated the dose can be escalated to a maximum of two tablets three times a day. The uptitration is usually attempted every 2-4 weeks, although rapid titration of 3-5 days has been suggested – it may not be well tolerated by some patients. As an alternative strategy, ISDN 20 mg tablet and hydralazine 25 mg tablet may be prescribed three times daily. These doses can be progressively increased to attain a dose similar to those used in FDC trial.
Adverse effects
Patient compliance to combination therapy is generally considered sub optimal due to high pill burden as well as frequent adverse effects. Common adverse reactions include headache, dizziness, hypotension, nausea, vomiting, tachycardia and asthenia. Slow up-titration of doses is recommended to promote tolerance and reduce occurrence of these events. Hydralazine has also been associated with peripheral neuritis and drug induced systemic lupus erythematosus (SLE) syndrome.
Cardiac Glycosides
In 1785, William Withering, an English botanist chemist and physician was the first person to publish a monograph describing that digitalis purpurea, a derivative of the purple foxglove flower was useful for the treatment of cardiac ‘dropsy’ or congestive cardiac failure. Foxgloves contain several cardiac glycosides whose basic chemical structure consists of three components: a sugar moiety, a steroid nucleus and a lactone ring. While the lactone ring is essential for inotropic activity, the remaining parts of the molecule primarily determine the potency and pharmacokinetics. Therapeutically the key cardiac glycoside is digoxin.
Endogenous cardiotonic steroids or digitalis-like factors have been subject of considerable interest for more than 50 years and have been proposed to play crucial role in regulation of renal sodium transport and arterial pressure. More recently they have been implicated in regulation of cell growth, differentiation, apoptosis, fibrosis, the modulation of immune functions and the control of several central nervous functions as well as carbohydrate metabolism.
Mechanism of action:
Digoxin has several direct as well as indirect effects on cardiovascular system with both desirable as well as undesirable consequences. Its beneficial effects in heart failure patients are generally ascribed to a positive inotropic effect, suppression of rapid ventricular rate in CHF associated atrial fibrillation and regulation of sympathetic nervous system overactivity.
At molecular level, digoxin inhibits sarcolemmal Na+/K+-ATPase or the sodium pump in cardiac myocytes. It binds and inhibits the phosphorylated α subunit of the Na+-K+-ATPase resulting in decreased extrusion of sodium and consequently its increased intracellular levels. This results in decreased extrusion of Ca2+ via the Na+/ Ca2+ exchanger leading to increased accumulation of calcium in sarcoplasmic reticulum by SERCA2. Consequently, increased amount of Ca2+ is released by each action potential leading to augmented myocardial contractility.
The electrophysiological cardiac effects of digoxin result from both direct as well as autonomic actions. At therapeutic concentrations, digoxin increases vagal outflow and inhibits sympathetic nervous system activity. These actions lead to decreased cardiac automaticity and increased maximal diastolic resting membrane potential in the atrial and atrioventricular nodal tissues. Digoxin has also been shown to prolong the effective refractory period and reduce conduction velocity in the AV nodal tissues. These cholinomimetic effects underlie its usefulness in patients with atrial fibrillation. However, toxic concentrations can disturb sinus rhythm and can progress to high grade AV block. At high concentrations, cardiac glycosides can increase sympathetic activity and predispose patients to atrial and ventricular arrhythmias by enhancing cardiac automaticity. As toxicity progresses, delayed after-depolarizations appear due to increased intracellular calcium loading and fluctuating free calcium ion concentrations. Initially these result into coupled beats or bigeminy followed by ventricular tachycardia and eventually ventricular fibrillations.
Pharmacokinetics:
Digoxin is administered orally and intravenously, in urgent cases. The elimination half life of digoxin is 36-48 hours in patients with normal renal function allowing once daily dosing and steady state blood levels are achieved in approximately a week after initiation of therapy. Digoxin is a polar compound and is eliminated primarily by kidneys. Thus increase in cardiac output and renal blood flow from the use of vasodilators or sympathomimetics may warrant adjustment of maintenance doses. Digoxin has a large volume of distribution due to its affinity for skeletal muscle and thus it is not effectively cleared by hemodialysis. Dose reduction is needed in elderly as well as in patients with chronic renal failure patients. Renal clearance of digoxin involves P-glycoprotein and thus clinically significant drug interactions can be observed with other cardiovascular agents such as verapamil, amiodarone and spironolactone. Determination of digoxin concentrations should be carried out by blood sampling after more than six hours of an oral dose or just before the next dose i.e. trough levels, to allow for its tissue distribution. The usual therapeutic range is considered to be 1–2 ng/ml, although toxicity has been observed at less than 1.5ng/mL in some patients.
Clinical Use:
Several placebo controlled studies have demonstrated that digoxin used for 1-3 months in patients with mild to moderate CHF can improve symptoms, quality of life and exercise tolerance irrespective of the underlying cardiac rhythm, cause of failure as well as concomitant therapies (with or without ACE inhibitors). The results of the Digitalis Investigation Group (DIG) trial demonstrated that in patients with NYHA class II or III CHF, treatment with digoxin for 2-5 years had no effect on mortality but modestly reduced the combined risk of death and hospitalization. A recent meta-analysis investigating the safety and efficacy of digoxin based on observational studies as well as trial data, reviewed 52 studies and concluded that digoxin has a neutral impact on mortality in randomized clinical trials and reduced rates of hospitalizations across all included study types.
Digoxin use in pharmacotherapy of heart failure is usually limited to two situations: in patients with heart failure and supraventricular tachyarrhythmias such as atrial fibrillation and CHF patients in normal sinus rhythm with persistent symptoms despite therapy with maximum tolerated doses of ACE inhibitors,beta blockers and aldosterone receptor antagonists. If a patient is currently on digoxin but not an ACE inhibitor or a beta blocker, treatment with digoxin need not be withdrawn, but therapy with suitable neurohormonal antagonists should be initiated. Treatment with digoxin is usually initiated and maintained at a dose of 0.125-0.25 mg per day. Low doses of 0.125 mg daily or alternate day is recommended in elderly patients (>70 years of age), patients with impaired kidney function or those with low lean body mass. Loading doses of digoxin are usually not required to initiate therapy in CHF patients.
Rapid digoxin loading is usually indicated in patients requiring control of ventricular rate in atrial fibrillation/ flutter and when alternative therapies have failed or are contraindicated. For intravenous loading, a dose of 0.25-0.5 mg is administered over several minutes, followed by 0.25 mg – 6 hourly to achieve a total loading dose of 0.75 – 1.5 mg. When used orally, digoxin loading is initiated with 0.5 mg dose followed by 0.25 mg – 6 hourly to achieve a similar total loading dose as above.
Adverse effects:
When carefully administered taking into account the drug and patient factors, digoxin is usually well tolerated in most individuals. The principal side effects include cardiac arrhythmias viz. ectopic and re-entrant cardiac arrhythmias and AV block and non cardiac effects including nausea, vomiting, diarrhoea, anorexia, disorientation, hallucinations and visual disturbances. Hypokalemia, hypocalcemia, hypomagnesemia and hypothyroidism increase the likelihood of cardiac toxicity of digoxin. Although overt digoxin toxicity is usually associated with serum digoxin levels of more than 2 ng/ml, symptoms may occur at lower concentrations if predisposing conditions are present. Usual treatment of digoxin toxicity includes drug withdrawal or dose reduction and treatment of cardiac arrhythmias and correction of electrolyte abnormalities. Patients at increased risk of toxic manifestations include elderly, those with renal function impairment, low lean body mass and those taking interacting drugs. In cases of life-threatening toxicity, anti-digoxin immunotherapy should be administered. This comprises of purified Fab fragments derived from specific anti-digoxin antibodies raised in sheep.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Pharmacology"
Pharmacology involves the study of drugs and how they affect the body. A pharmacologist contributes to drug development by researching and testing how the body reacts to medication, and whether the medication can have a positive impact on the body in terms of fighting illness and disease.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: