Pharmaceutical Cocrystals and Co-amorphous systems: Chemistry, Solubility and Performance
Info: 3242 words (13 pages) Dissertation
Published: 9th Dec 2019
Tagged: ChemistryPharmacology
Pharmaceutical Cocrystals and Co-amorphous systems: Chemistry, Solubility and Performance
Introduction
More than 75% of drug candidates and 40% of marketed drugs are poorly soluble in aqueous buffer systems. A drug’s aqueous solubility is a profoundly important factor in drug discovery and development. Drugs with low solubility and dissolution rate have limited and variable absorption and bioavailability [2]. Various techniques are used for the improvement of the solubility and dissolution rate of drugs depending on drug properties, including: cosolvency, using surface active agents, pH control, salt formation, complexing agents, hydrotropism, particle size reduction or nanosizing, prodrugs, solid dispersions, lipid-based formulations, inorganic matrices , and crystal engineering. Crystal engineering is one of the classical approaches to change the physicochemical properties of pharmaceuticals. Cocrystals are single crystalline phase materials composed of two compounds (drug + conformer) via noncovalent interactions in certain stoichiometric ratios. They are typically formed via strong hydrogen bonds between carboxylic acids, amides, carbohydrates, alcohols, and amino acids [2, 3]. This method against of salt formation is not limited to ionizable compounds. In addition to the advantages of cocrystals, co-amorphous or amorphous cocrystal can improve stability (solution and physical stability) of amorphous form of drugs. Although amorphous systems are a useful way of improving solubility and dissolution rate, they are thermodynamically unstable and will crystallize over pharmaceutically relevant time scales. Excipients such as polymers or amino acids can be used to stabilize amorphous systems [4]. An overview of cocrystals and co-amorphous pharmaceuticals, preparation and characterization methods, and solubility and dissolution properties is given in the following paragraphs.
Crystal engineering
Crystal engineering is a technique in which intermolecular interactions in the context of crystal packing is investigated in order to design new solids with desired physicochemical properties. The challenges of low aqueous solubility provide an ideal situation for the application of crystal engineering technique for improving bioavailability and developing stable and robust pharmaceutical products [2]. This technique has profound advancement over the last 50 years because the common way to deliver drugs is solid dosage forms. Moreover, crystalline forms are more stable, reproducible, and amenable to purify compared to other types of solid forms, so the selection of a specific crystal form is a critical parameter. Traditional crystal engineering methods are limited to polymorphs, salts, solvates or hydrates [5]. Polymorphism is the ability of a drug to exist in more than one form or crystal structure. Crystallization of a solid-state structure containing solvent molecules is named solvate, and if the solvent is water, the crystalline structure is named hydrate. Salt formation is another classic approach to alter the physicochemical properties of drugs. Recently, liquid form of salts (ionic liquid) has been considered as alternative method to enhance solubility. Nevertheless, a drug should have ionizable functional groups for salt formation[6]. Therefore, cocrystals are considered as applicable crystal engineering method in pharmaceutical sciences.
What is a cocrystal?
Generally, a co-crystal is a multicomponent molecular crystal; that is, a crystalline substance composed of two or more chemically different molecules. However, this definition can also include solvates, hydrates, and both stoichiometric and non-stoichiometric lattice inclusion compounds. Therefore, a strict definition, which indicates the practical difference between cocrystals and its other subcategories, would be a crystalline substance that includes a solid coformer rather than water (hydrates) or a solvent (solvates) that might have been included accidentally as a potential solvent for the active pharmaceutical ingredient. Furthermore, it is also accepted that cocrystals composed of opposite enantiomers of the same substance cannot be regarded as co-crystals, since both enantiomers can be related to each other within the crystal by inversion or improper rotation. In Pharmaceutical cocrystals, at least one of the components is an API and the cocrystal former has to be non-toxic with no side effects and ideally be included on the US FDA ‘Everything Added to Food in the United States’ (EAFUS) list[7].
Why Cocrystals?
The growing interest in the use of cocrystals arises from the fact that differences in physicochemical properties such as habit, bulk, density, solubility, compressibility, friability, melting point, hygroscopy, and dissolution rate, offer a wide range of solid forms that are available for formulation. The main advantage of cocrystals compared to salts, which is one of the preferred approaches to increase aqueous solubility, is their applications for drugs with non-ionizable groups. In addition, cocrystals are superior to amorphous form of drugs because of their easier and more reproducible characterization, lower hygroscopicity, and greater stability [8]. They are applied in pharmaceutical sciences to modulate stability, physicochemical, and pharmacokinetic properties. However, the importance of application of cocrystals is improving the solubility and dissolution rate of poorly soluble APIs [9, 10].
Cocrystals structure and preparation:
Three steps are involved in designing cocrystals: conformer selection, computational analysis and characterization of the cocrystals. Typically, coformers like carboxylic acids, amides and alcohols which have the ability to form hydrogen bond with API, are preferred, as hydrogen bond formation is the most common interaction between co-crystal components. The synthon is a model which is representative of the entire crystal structure and when the crystal pattern is repeated regularly, the pattern is called super molecular synthon. Supermolecular Homosynthons are composed of identical self-complementary functionalities, like acid-acid, while Supermolecular Heterosynhon include different but complementary functionalities, like acid-amide[11]. Since the first cocrystals were discovered by dry grinding and all the components are solid under ambient conditions, it has been known that mechanochemistry (solid state methods) can be utilized to produce cocrystals through solvent-free or solvent-reduced methods, but it has recently been realized that ‘solvent-drop’ or ‘liquid assisted’ grinding are preferred methodologies. Solvent evaporation, Slurry crystallization technique, wet milling of the solid components, melting and cooling are the other methods of preparation of cocrystals[10].
Cocrystal solubility:
Cocrystal solubility is usually determined by kinetic or thermodynamic solubility approaches. Kinetic solubility shows cocrystals dissolution rate and indicates non-equilibrium solubility (apparent solubility) due to the cocrystal instability in solution, and a slight excess of coformer in the structure of synthesized cocrystal[12]. Thermodynamic solubility or equilibrium solubility of a cocrystal depends on drug and coformer concentration in the solution phase; when the cocrystal is not stable in solution, thus dissociating into its components in solution. In this state, the mass of excess solid phase is an important aspect that should be considered. To put it another way, thermodynamic solubility provides information about the origin of cocrystal solution phase behavior, which is necessary for addressing oral absorption limitations of poorly soluble drugs. It can be calculated using the proposed method by Rodríguez-Hornedo and coworkers, where transition points (eutectic concentration) of each component are determined at the three-phase equilibrium (drug, cocrystal and solution phase). The concentrations at eutectic point give information about cocrystal stability which is directly related to cocrystal solubility [13, 14].
Characterization of cocrystals:
Cocrystal formation can be deducted from the results of several techniques used for characterization of crystalline molecular solids.
1. Single X-ray crystallography
Single-crystal X-ray diffraction is an analytical technique which provides detailed information about the internal lattice of crystalline substances, including unit cell dimensions, bond-lengths, bond-angles, and details of site-ordering. This method uniquely determines the crystal form and a full 3D representation of structure[7].
2. PXRD
Powder X-ray diffraction (PXRD) is a rapid analytical technique primarily used for phase identification of a crystalline material and provides information on unit cell dimensions. PXRD is most widely used for the identification of unknown crystalline materials[7].
3. Fourier transform infrared spectroscopy (FT-IR)
Fourier transform infrared spectroscopy (FT-IR) is a powerful and sensitive tool for solid-state studies which all spectral bands can be assigned to specific features of the molecule under investigation and identify molecular interactions (such as hydrogen bonding) between two functional groups of a single (polymorphism or conversion of crystalline to amorphous form) or two components (cocrystal and co-amorphous) [7].
4. Differential scanning calorimetry
Differential scanning calorimetry (DSC) is the most commonly used thermal analysis method. It is a useful technique to determine important melting point, glass transition temperatures and enthalpy of fusion of crystalline materials. Physicochemical properties such as the transition temperature (i.e., to determine enantiotropic/monotropic relationships between polymorphic forms) or melting temperature can be easily observed as an exothermic/endothermic event in the DSC thermograms. Analysis of an amorphous phase using DSC should reveal a glass transition temperature (Tg) and/or a crystallization temperature (without endothermic melting peaks), whereas melting temperature would be expected for a crystalline material. DSC can be used in cocrystal studies to identify cocrystal formation between two components with appearance of new endothermic peak in the thermogram[15].
Solid dispersions:
As shown in figure 1, co-amorphous formulations are a class of glass solutions which itself is a subcategory of solid dispersions. Dispersion of one or more active ingredients in an inert carrier at the solid state prepared by the melting, solvent, or melting-solvent method is the common definition of solid dispersions. Glass solutions (amorphous) are single phase amorphous systems, which are considered as supersaturated drug delivery systems, capable of increasing absorption and bioavailability of drugs. However, Because of their short-range molecular orders and lack of well-defined molecular conformation, amorphous solids have high mobility and internal energy, thus leading to thermodynamic instability. In order to both achieve the primary advantage of amorphous solutions and overcome their disadvantage, polymeric amorphous solid dispersions are preferred, in which the drug is incorporated as molecular dispersion in a glass polymeric matrix, stabilized by physical separation of the molecules inside the polymer chains. Due to the challenges in developing polymeric amorphous solid dispersion, such as low solubility of drug in the polymer resulting in incorporation of high amount of volume and increasing the final volume of dosage forms plus sensitivity to heat and humidity due to the hygroscopicity of the polymers, their usage in pharmaceutical industry are limited [1, 4].
Co-amorphous pharmaceuticals:
Co-amorphous solid dispersions are the alternative to polymeric amorphous solid dispersions. Co-amorphous solid dispersions are defined as a multi-component single phase amorphous solid system which lacks recurrence in lattice and is associated by weak and discrete intermolecular interactions between the components. Similar to amorphous system of single component systems, co-amorphous systems contain short-range interactions such as hydrogen bonding of carboxylic acids, phenols/alcohols. As the components in Co-amorphous systems have low MW, the amount of stabilizing excipient can be significantly reduced. The two common types of co-amorphous solid dispersions are drug–drug combinations, in which two pharmacologically relevant drugs intended for multidrug therapies are combined, while both drugs stabilize each other, both of the poorly soluble drugs achieve a higher solubility and dissolution rate. and the second type is the drug–excipient mixtures, where a low molecular weight excipient such as amino acids are used to provide a stable and quick-dissolving mixture. The first drug-drug CAM was cimetidine-naproxen co-amorphous which was reported by Yamamura et al., in 1996.Nonetheless, there are limited number of drug-drug CAM reported, since it is not easy to find two pharmacologically related drug pairs that are also able to form glass solution in the required doses. In contrast, combinations of drugs with inert molecules, capable of forming hydrogen bonding with low toxicity, such as carboxylic acids (citrate, tartrate) weak bases (meglumine, flavonoids) and amino acids is common and of more interest[4, 16].
Co-amorphous preparation:
Depending on the physicochemical properties of the drug and excipients, different preparation methods can be used to prepare co-amorphous solids. Laboratory scale preparative techniques include, quenching, solvent evaporation and ball milling, which are ideal for screening purposes. Furthermore, freeze drying, spray drying, and ultrasound extrusion are reported as large-scale preparative methods for co-amorphous formulations[16].
Solubility and dissolution properties of Co-amorphous pharmaceuticals:
As mentioned above, higher internal energy of amorphous phases results in higher solubility and dissolution rate. Furthermore, it has been shown that co-amorphous systems have improved solubility and dissolution rate over both their crystalline forms and their individual amorphous forms. For instance, in case of naproxen-cimetidine co-amorphous, there is a 2-fold change in dissolution rate of cimetidine in co-amorphous form, compared to its pure amorphous and crystalline form, which show the similar dissolution rate due to recrystallization of amorphous form. In fact, not only dissolution rate of the drug can be increased by co-amorphization, but also solvent-induced recrystallization upon dissolution can be prevented and molecular interactions have a crucial role in this stabilization process[8].
Concluding remarks:
Co-crystal formation provides huge scope for controlled modification of important pharmaceutical properties such as habit, density, solubility, compressibility, melting point, hygroscopy, and dissolution rate. Although amorphous drugs and cocrystals might show similar solubility and dissolution profile, yet cocrystals have some advantages over co-amorphous. Due to their crystalline nature, cocrystals are more stable to humidity and storage, drug processing, wet granulation, tableting, compaction, etc. Plus, the possibility of undergoing phase transformations is low in cocrystals compared to co-amorphous solids[7].
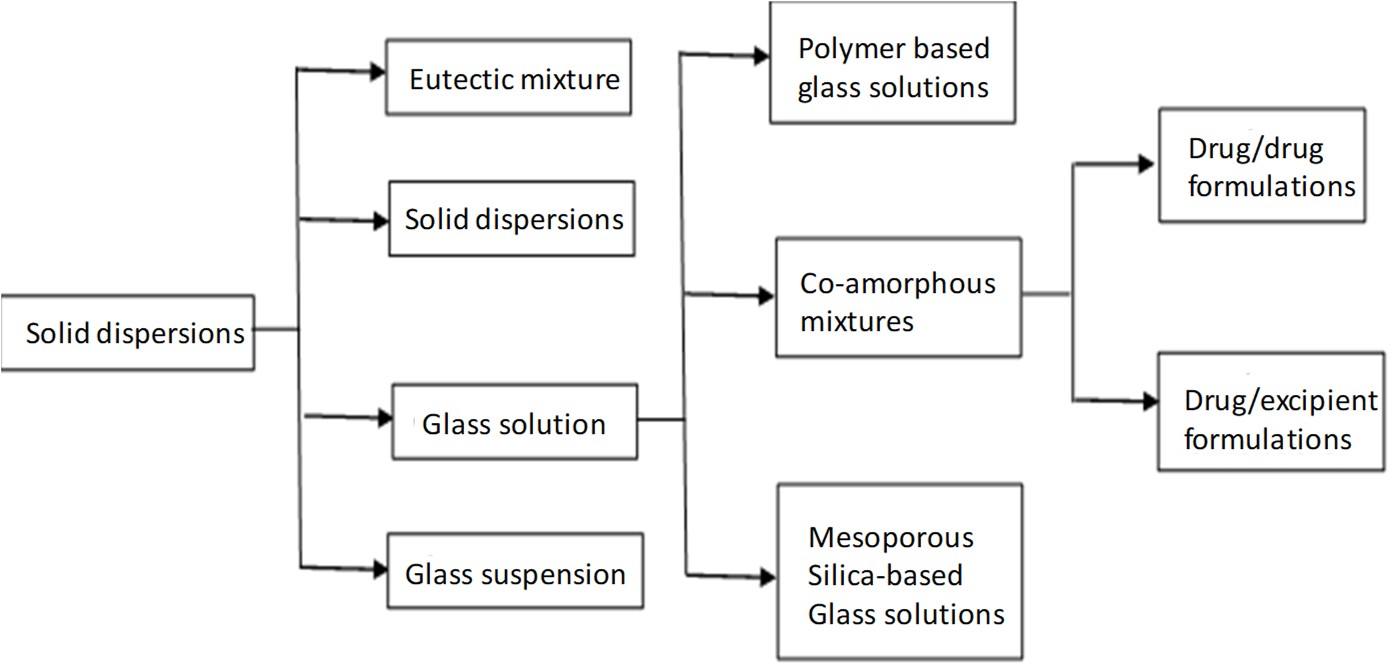
Figure1
S.J. Dengale et al. / Advanced Drug Delivery Reviews 100 (2016) 116–125
S.J. Dengale et al. / Advanced Drug Delivery Reviews 100 (2016) 116–125
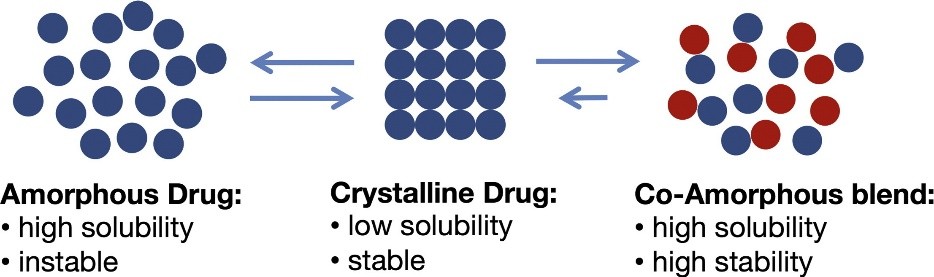
Figure 2
Karagianni, A., et al.
References:
1. Karagianni, A., K. Kachrimanis, and I.J.P. Nikolakakis, Co-amorphous solid dispersions for solubility and absorption improvement of drugs: Composition, preparation, characterization and formulations for oral delivery. 2018. 10(3): p. 98.
2. Blagden, N., et al., Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Advanced Drug Delivery Reviews, 2007. 59(7): p. 617-630.
3. Kawakami, K.J.A.d.d.r., Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. 2012. 64(6): p. 480-495.
4. Chavan, R.B., et al., Co amorphous systems: A product development perspective. International Journal of Pharmaceutics, 2016. 515(1): p. 403-415.
5. Desiraju, G.R.J.J.o.t.A.C.S., Crystal Engineering: From Molecule to Crystal. 2013. 135(27): p. 9952-9967.
6. Serajuddin, A.T.J.A.d.d.r., Salt formation to improve drug solubility. 2007. 59(7): p. 603-616.
7. Steed, J.W., The role of co-crystals in pharmaceutical design. Trends in Pharmacological Sciences, 2013. 34(3): p. 185-193.
8. Babu, N.J. and A. Nangia, Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Crystal Growth & Design, 2011. 11(7): p. 2662-2679.
9. Schultheiss, N., A.J.C.g. Newman, and design, Pharmaceutical cocrystals and their physicochemical properties. 2009. 9(6): p. 2950-2967.
10. Shan, N. and M.J.J.D.d.t. Zaworotko, The role of cocrystals in pharmaceutical science. 2008. 13(9-10): p. 440-446.
11. Bavishi, D.D. and C.H. Borkhataria, Spring and parachute: How cocrystals enhance solubility. Progress in Crystal Growth and Characterization of Materials, 2016. 62(3): p. 1-8.
12. Thakuria, R., et al., Pharmaceutical cocrystals and poorly soluble drugs. 2013. 453(1): p. 101-125.
13. Good, D.J., N.J.C.G. Rodriguez-Hornedo, and Design, Solubility advantage of pharmaceutical cocrystals. 2009. 9(5): p. 2252-2264.
14. Keramatnia, F., A. Shayanfar, and A.J.J.o.p.s. Jouyban, Thermodynamic Solubility Profile of Carbamazepine–Cinnamic Acid Cocrystal at Different p H. 2015. 104(8): p. 2559-2565.
15. Lu, E., N. Rodríguez-Hornedo, and R.J.C. Suryanarayanan, A rapid thermal method for cocrystal screening. 2008. 10(6): p. 665-668.
16. Dengale, S.J., et al., Recent advances in co-amorphous drug formulations. Advanced drug delivery reviews, 2016. 100: p. 116-125.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Pharmacology"
Pharmacology involves the study of drugs and how they affect the body. A pharmacologist contributes to drug development by researching and testing how the body reacts to medication, and whether the medication can have a positive impact on the body in terms of fighting illness and disease.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: