Regulation of the Cd36 Scavanger Receptor by the Antioxidant 7,8-dihydroneopterin
Info: 41338 words (165 pages) Dissertation
Published: 14th Feb 2022
Tagged: Biology
ABSTRACT
OxLDL uptake via the CD36 scavenger receptor leads to foam cell formation and is at the core of the development of atherosclerosis. One potential protective mechanism against this process involves the human macrophage derived anti-oxidant 7,8-dihydroneopterin (7,8-NP) down regulating CD36. This study characterised CD36 down regulation in the U937 monocyte-like cell line, and examined a mechanism of action involving MAP kinase mediated control of the PPARYγ transcription factor.
Western blot analysis showed that it in U937 cells 7,8-NP concentrations up to 150µM down regulated CD36 to ~40% of basal levels over 24 hours. The effect seen here was stronger than that previously observed with human monocyte derived macrophages (HMDM). The oxidised product of 7,8-NP, neopterin, had no significant effect. CD36 levels were able to recover after down regulation and neared control levels 24 hours after 7,8-NP removal.
CD36 protein levels were found to be under control of PPARγ and it was shown that 7,8-NP likely only has its effect at the transcriptional level, and did not enhance the proteolytic removal of the CD36. PPARγ contains a MAP kinase binding site which when phosphorylated prevents the transcription of CD36. Co-incubation of selective MAP kinase inhibitors SP600125 (JNK) and PD98059 (ERK1/2) with 7,8-NP failed to block CD36 down regulation. The effect of p38 and NF-κB signalling in CD36 down regulation was additionally explored using their inhibitors (SB202190 and BAY 11-7082 respectively), but likewise did not block 7,8-NP’s effect.
The results confirm that in the U937 cell line 7,8-NP can decrease the levels of CD36, and that a regulatory pathway involving PPARγ is likely. It is also shown that 7,8-NPs mechanism of action does not involve the activation of a MAP kinase cascade phosphorylating PPARγ. This points towards a different mechanism of action, possibly involving PPARγ’s lipid ligand binding site.
TABLE OF CONTENTS
Click to expand Table of Contents
1. INTRODUCTION
1.1 Overview
1.2 Atherosclerosis
1.2.1 Atherosclerosis and cardiovascular disease
1.2.2 The stages of plaque progression
1.2.3 Lipoproteins and oxidised LDL
1.2.4 Macrophages, foam cell formation and cell death
1.3 Plaque initiation theories
1.3.1 Response to injury theory
1.3.2 Response to retention theory
1.3.3 Oxidative modification hypothesis
1.4 Scavenger receptors
1.4.1 Scavenger receptors
1.4.2 CD36
1.4.3 CD36 structure
1.4.4 CD36 in atherosclerosis
1.5 7,8-Dihydroneopterin
1.5.1 GTP, 7,8-Dihydydroneopterin and products
1.5.3 Antioxidant down regulation of CD36
1.5.4 Other modes of CD36 down regulation
1.6 Peroxisome proliferator activated receptor-ɣ
1.6.1 PPARγ structure and function
1.6.2 Modulation of PPARγ activity
1.7. MAP kinases
1.7.1 ERK cascade
1.7.2 JNK cascade
1.7.3 P38 cascade
1.7.4 MAP kinases & atherosclerosis
1.8 Objective of research
2. MATERIALS & METHODS
2.1 Reagents, media and buffers
2.1.1 Reagents
2.1.2 Media
2.1.3 General solutions and buffers
A. Roswell Park Memorial Institute (RPMI)-1640 media (with or without phenol red)
B. Phosphate buffered saline (PBS)
C. 7,8-dihydroneopterin solution
D. D-neopterin solution
E. Triton lysis solution
F. MOPS buffer for SDS-PAGE
G. Transfer buffer for western blotting
H. Tris-buffered saline (TBS)
I. TBSM
J. Cracker buffer for SDS-PAGE
K. Restore stripping buffer
L. Ponceau S stain
M. Propidium iodide solution
N. Phorbol 12-myristate 12-acetate (PMA)
2.2 Methods
2.2.1 Cell culture based experiments
2.2.2 Cell viability assays
2.2.3 Determination of protein concentration
2.2.4 SDS-PAGE
2.2.5 Western blot analysis
2.2.6 Flow cytometry determination of U937 CD36 cell surface expression
2.2.7 Statistical analysis
3. RESULTS
3.1 Characterising the expression of CD36
3.1.1 The U937 cell line produces detectable basal levels of CD36
3.1.2 Triton X-100 as a lysing agent
3.1.3 Effect of PMA of U937 expression
3.2 Characterization of CD36 down regulation via 7,8-NP
3.2.1 7,8-NP reduces the expression of the CD36 scavenger receptor in the U937 cell Line
3.2.2 7,8-NP induced down regulation occurs in both adherent and suspension well plates
3.2.3 7,8-NP down regulation can be observed using both wet and semi-dry protein transfer protocols
3.2.4 Time course of 7,8-NP induced CD36 down regulation
3.2.5 Recovery of CD36 expression after 7,8-NP removal
3.2.6 Cellular viability of the recovery experiment
3.2.7 Effect of neopterin on CD36 expression in the U937 cell line
3.3 Transcriptional control of CD36
3.3.1 Effect of PPARγ inhibitor GW9662 on CD36 expression
3.3.2 Effect of GW9662 on cellular viability
3.3.3 Effect of GW9662 in conjunction with 7,8-NP on CD36 expression
3.3.4 Effect of increasing concentrations of cycloheximide on cellular viability
3.3.5 Effect of cycloheximide on CD36 expression
3.3.6 Cellular viability of the cycloheximide experiment
3.4 Effect of MAP kinase inhibitors on CD36 down regulation
3.4.1 Effect of JNK inhibition via SP600125 on 7,8-NP induced down regulation in the U937 cell line
3.4.2 Effect of JNK inhibition on cellular viability
3.4.3 Effect of MEK inhibition via PD98059 on 7,8-NP induced down regulation in the U937 cell line
3.4.4 Effect of MEK inhibition on cellular viability
3.4.5 Effect of p38 inhibition via SB202190 on 7,8-NP induced down regulation in the U937 cell line
3.4.6 Effect of p38 inhibition on cellular viability
3.4.7 Effect of NF-kB inhibition via BAY 11-7082 on 7,8-NP induced down regulation in the U937 cell line
3.4.8 Effect of NF-kB Inhibition on cellular viability
3.5 CD36 flow cytometry
3.5.1 CD36 flow cytometry protocol design
3.5.2 Down regulation of CD36 detected via flow cytometry
4. DISCUSSION
4.1 Characterising CD36 down regulation
4.2 CD36 Down Regulation Measured via Flow Cytometry
4.3 Transcriptional Control of CD36
4.4 The Effect of MAP Kinase Inhibitors
4.4.1 JNK Inhibition
4.4.2 ERK1/2 Inhibition
4.4.3 p38 Inhibition
4.4.4 NF-κB Inhibition
4.5 A different mechanism of effect
4.6 Incomplete Down regulation
4.7 Future Research
4.8 Summary
BIBLIOGRAPHY
LIST OF FIGURES
INTRODUCTION
Figure 1.1 The development of an atherosclerotic plaque
Figure 1.2 The formation of a lipid laden foam
Figure 1.3 The structure of CD36
Figure 1.4 The biosynthetic pathway of 7,8-NP and its reduced products
Figure 1.5 Protective mechanisms of 7,8-NP
Figure 1.6 Regulation mechanics of PPARγ
RESULTS
Figure 3.1.1 The U937 cell line produces detectable basal levels of CD36
Figure 3.1.2 0.5% TritonX-100 effectively lysis cells
Figure 3.1.3 PMA has a limited effect on U937 expression
Figure 3.2.1 7,8-NP reduces the expression of the CD36 scavenger receptor in the U937 cell line
Figure 3.2.2 7,8-NP down regulation can be observed using both adherent and suspension plates
Figure 3.2.3 7,8-NP down regulation can be observed using both wet and semi-dry transfer protocols
Figure 3.2.4 7,8-NP down regulates CD36 over 24 hours
Figure 3.2.5 CD36 expression recovers 24 hours after 7,8-NP removal
Figure 3.2.6 CD36 expression recovers 24 hours after 7,8-NP removal
Figure 3.2.7 Neopterin has a limited effect on CD36 expression
Figure 3.2.8 Images of U937 cells after treatment with increasing concentrations of neopterin
Figure 3.3.1 PPARγ inhibitor GW9662 decreases CD36 expression
Figure 3.3.2 Effect of GW9662 on cellular viability using propidium iodide
Figure 3.3.3 GW9662 and 7,8-NP do not have an additive effect on 7,8-NP expression
Figure 3.3.4 Effect of increasing concentrations of cycloheximide on cellular viability
Figure 3.3.5 Effect of cycloheximide on CD36 expression
Figure 3.3.6 Effect of cycloheximide and cycloheximide+7,8-NP on U937 cell viability using trypan blue
Figure 3.4.1 JNK inhibition via SP600125 does not alleviate CD36 down regulation
Figure 3.4.2 Effect of SP600125 on U937 cell viability using propidium iodide
Figure 3.4.3 MEK inhibition via PD98059 does not alleviate CD36 down regulation
Figure 3.4.4 Effect of PD98059 on U937 cell viability using propidium iodide
Figure 3.4.5 p38 inhibition via SB202190 does not alleviate CD36 down regulation
Figure 3.4.6 Effect of SB202190 on U937 cell viability using propidium iodide
Figure 3.4.7 NF-kB inhibition via BAY 11-7082 does not alleviate CD36 down regulation
Figure 3.4.8 Effect of BAY 11-7082 on U937 cell viability using propidium iodide
Figure 3.5.1.1 Flow cytometer traces for CD36 antibody on U937 cells
Figure 3.5.1.2 Use of the flow cytometry CD36 antibody on the U937 cell line
Figure 3.5.1.2 Flow cytometer traces for increasing concentrations of 7,8-NP on U937 cells
Figure 3.5.2.2 Effect of 7,8-NP on U937 cells detected using flow cytometry
ABBREVIATIONS
| 7,8-NP | 7,8-Dihydroneopterin |
| AAPH | 2,2′-Azobis(2-amidinopropane) dihydrochloride |
| ApoB100 | Apoplipoprotein B100 |
| BCA | Bicinchoninc acid |
| BMM | Bone marrow-derived macrophage |
| BSA | Bovine serum albumin |
| CD36 | Cluster of differentiation 36 |
| CE | Cholesterol Esters |
| DMSO | Dimethyl sulphoxide |
| DNA | Deoxyribonucleic acid |
| EDTA | Ethylenediaminetetraacetic acid |
| ERK | extracellular signal-regulated kinase |
| FL | Fluorescence filter |
| FRB | Free Radical Biochemsitry |
| FSC | Forward Scatter |
| GAG | Glycosaminoglycan |
| GCH-1 | GTP cyclohydrolase-I |
| GTP | Guanosine triphosphate |
| H2O2 | Hydrogen peroxide |
| HCl | Hydrochloric acid |
| HDL | High density lipoprotein |
| HETE | Hydroxyeicosatetraenoic acid |
| HMDM | Human monocyte derived macrophage |
| HOCL | Hypochlorous acid |
| HODE | Hydroxyoctadecadienoic acid |
| HRP | Hydrogen peroxidase |
| IL | Interleukin |
| INF-γ | Interferon-γ |
| JNk | c-Jun N-terminal kianse |
| LDL | Low-density lipoprotein |
| LOX-1 | Lectin-like oxidized low-density lipoprotein receptor-1 |
| LPS | Lipopolysaccharide |
| MAP | Mitogen activated protein |
| MAPKAPK | MAPK- activated protein kianse |
| MAPKK | Map kinase kinase |
| MAPKKK | Map kinase kinase kinase |
| MOPS | 4-Morpholine-propanesulfonic acid |
| MPM | Peritoneal macrophage |
| NaCl | Sodium chloride |
| NaCO3 | Sodium carbonate |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| NaHCO3 | Sodium bicarbonate |
| NaOH | Sodium hydroxide |
| oxLDL | Oxidatievly modified low-density lipoprotein |
| PBS | Phosphate buffered saline |
| PI | Propidium iodide |
| PKA | Protein Kianse A |
| PKB | Protein kinase b |
| PKC | Protein kinase c |
| PMA | Phorbol 12-myristate 12-acetate |
| PPARγ | Peroxisome proliferator-activated receptor-γ |
| PTPS | 6-pyruvonyltetrahydropterin synthase |
| ROS | Reactive oxygen species |
| RPMI-1640 | Roswell Park Memorial Institute 1640 |
| RXR | Retinoid X Receptor |
| SDS | Sodium dodecyl sulphate |
| SR-A | Scavanger Receptor A |
| STAT | Janus kinase-signal transducers and activators of transcription |
| TBS | Membrane blocking solution |
| TBS | Tris-buffered saline |
| TBSM | Membrane blocking solution + milk |
| TGF | Trasforming growth factor |
| TLR | Toll like receptor |
| TNFα | Tumor necrosis factor α |
| UV | Ultraviolte |
| XO | Xanthine Oxidase |
1. INTRODUCTION
1.1 Overview
Cardiovascular disease is a leading cause of death accounting for 16.7 million fatalities globally each year (Dahlöf, 2010). A major pathology of cardiovascular disease is atherosclerosis, otherwise known as the hardening of the artery wall (Hansson, 2005). This process is characterised by the recruitment of immune cells and the deposition of lipids alongside cellular debris within the arterial wall. Over time this leads to the formation of atherosclerotic plaques and results in the occlusion of blood flow (Lusis, 2000). Cardiovascular disease may manifest as angina, stroke or myocardial infarction (Roger et al., 2011).
Oxidative stress and inflammatory cell death are at the core of the development of atherosclerosis. One of the fundamental interactions during this process is the death of macrophage cells after exposure to oxidatively modified low density lipoprotein (oxLDL) (Moore & Tabas, 2011). Low density lipoprotein (LDL) is a key component of the lipoprotein pool that functions in the transportation of lipids around the body. Oxidative modification of this particle results in oxLDL which is pro-inflammatory and cytotoxic (Berliner & Heinecke, 1996). OxLDL will interact with and be taken up by macrophage cells leading to the formation of foam cells (Henriksen, Mahoney, & Steinberg, 1983). These foam cells remain within the artery wall and make up a significant part of the atherosclerotic plaque (Stocker & Keaney, 2004). The inflammatory properties of oxLDL will also contribute to higher levels of oxidative stress within the arterial wall and in doing so recruit more macrophages to this location (Park, Febbraio, & Silverstein, 2009).
The initial step that results in the uptake of oxLDL is mediated by a class of cell surface receptors termed scavenger receptors. Cluster of differentiation 36 (CD36) is the scavenger receptor most associated with oxLDL triggered cytotoxicity and oxLDL uptake as CD36 contains motifs on its extracellular portion that allows for recognition and binding of the oxidised components of oxLDL (Ohidar, 2010). Mechanisms that are able to decrease levels of CD36 are of increasing interest in order to slow the progression of atherosclerosis.
One potential mechanism for CD36 down regulation relates to the human macrophage derived anti-oxidant 7,8-dihydroneopterin (7,8-NP). The protective role of 7,8-NP against macrophage foam cell formation is believed to be two-fold; 1) Its antioxidant activity which scavenges any reactive oxygen species (ROS) that may be generated in response to oxLDL (Steven Gieseg, Crone, Flavall, & Amit, 2008; Steven Gieseg, Reibnegger, Wachter, & Esterbauer, 1995). 2) The down regulation of CD36 resulting in a decrease in the amount of oxLDL that is taken up by macrophages in the first place (S. Gieseg, Z. Amit, Y.-T. Yang, A. Shchepetkina, & H. Katouah, 2010).
This research focuses on the second mode of action and explores how 7,8-NP is able to down regulate CD36 expression. By testing the effects of various inhibitors of intracellular signalling, we explore the mechanism of CD36 down regulation by 7,8-NP, with the aim of increasing our understanding its protective effect in atherosclerosis. Specifically 7,8-NP’s ability to down regulate the CD36 scavenger receptor and thus prevent foam cell formation.
1.2 Atherosclerosis
1.2.1 Atherosclerosis and cardiovascular disease
Atherosclerosis is the underlying process in cardiovascular disease which includes coronary heart disease, strokes and peripheral vascular disease. Altogether cardiovascular disease accounts for 30% of deaths globally (Santulli, 2013). The progression of atherosclerosis is a chronic and complex process. Its development involves endothelial dysfunction, arterial inflammation, immune cell recruitment and the thickening of the arterial intima through a build-up of cellular debris, lipids, cholesterol and calcium deposition (Bobryshev, 2006). Remodelling of the arterial wall results in large occlusions known as atherosclerotic plaques that restricts blood flow and reduces oxygen supply. As an atherosclerotic plaque progresses the lesion can become unstable and may rupture. Once ruptured the plaque will release its contents into the blood stream triggering thrombus formation, which if lodged in the small capillary beds, can further restrict or completely block blood supply (Falk, 1983). The end result of this blockage may lead to myocardial or cerebral infarction depending on the location of the plaque (Hansson & Hermansson, 2011; Libby, Ridker, & Maseri, 2002). Many risk factors will influence the likelihood of developing atherosclerosis and its severity. These include an individual’s genetic predisposition, along with obesity, age, gender, cigarette and alcohol use, sedentary lifestyle, hypertension and the presence of type II diabetes and hyperlipidaemia (Libby et al., 2002; Meyer & Schmitt, 2000).
1.2.2 The stages of plaque progression
The formation of an atherosclerotic plaque develops over a lifetime and goes through several stages (Figure 1.1). Six stages of plaque progression have been identified as determined by plaque complexity (Ross, 1995). Plaques found in adolescents are often identified as being in stage 1 or 2 of development. Here monocyte infiltration into the intimal layer of the arterial wall, along with the presence of plasma-derived lipid, initiates the thickening of the arterial wall (Ross, 1986; Stary et al., 1995; D Steinberg, 1989). As this thickening expands the addition of further lipids and cells, along with cell death leads to the development of Type III plaques. Necrotic core formation may then occur in which a lipid laden area devoid of cells develops deep within the arterial wall. These plaques are classified as being Type IV and can also be characterized by a thin layer of tissue that separates the necrotic core from the lumen. This layer will develop into a thick fibrous cap in Type V plaques. The final stage of plaque formation involves a more complex structure in which calcium deposits and ulceration develop, along with atrial vascular remodelling (Ross, 1986; Stary et al., 1995; D Steinberg, 1989).
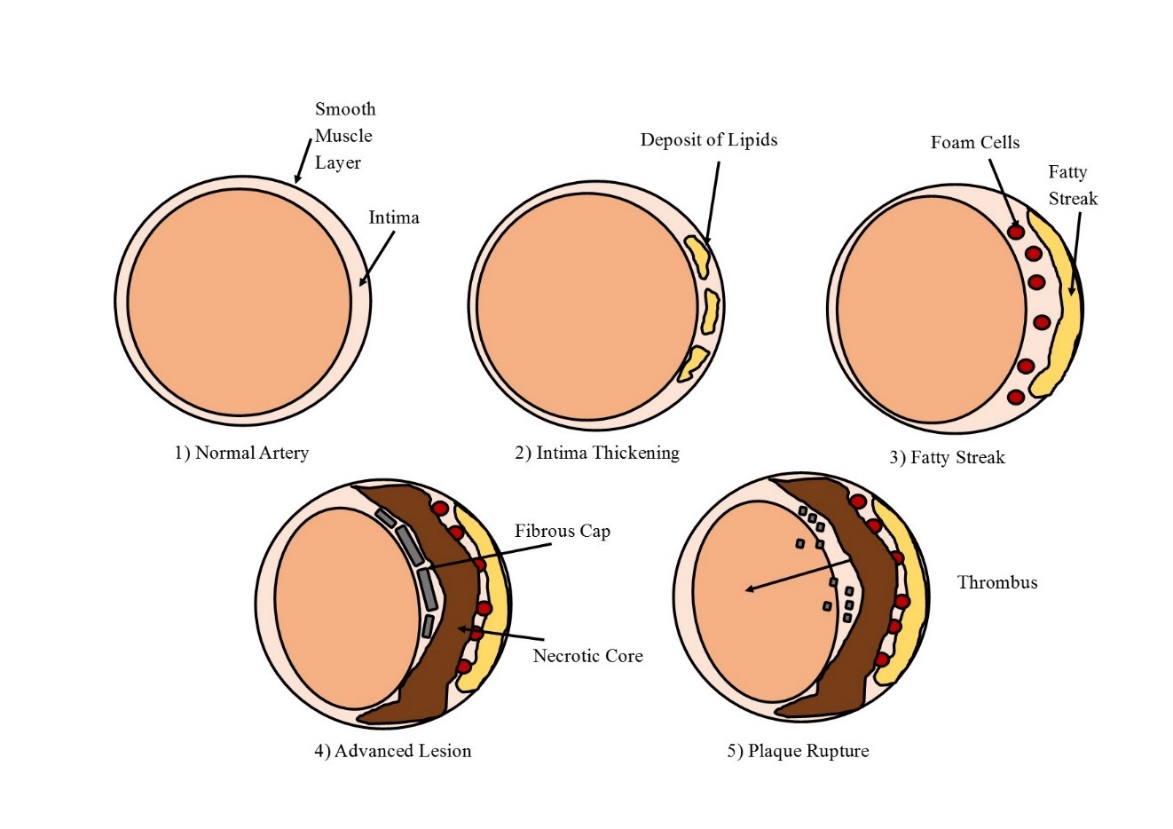
Figure 1. 1 The development of an atherosclerotic plaque
The various stages of atherosclerotic plaque development, depicting the normal artery (1), lipid deposition (2), fatty streak formation (3), necrotic core and fibrous cap (4) and plaque rupture forming a thrombus (5). Adapted from Cross, 2016.
1.2.3 Lipoproteins and oxidised LDL
Several theories have been proposed to explain the initial process that leads to the beginning of plaque formation including the “Response to injury”, “Response to retention” and the “Oxidative modification” hypothesis. A common theme in all these theories is the role that LDL and immune cells play in atherosclerotic progression.
The body’s lipoprotein pool is an important source of lipids, both in normal circulation and throughout atherosclerotic plaques (Lusis, 2000). Lipoproteins are not homogenous but rather are split into many sub-fractions based on size, density and apolipoprotein content. Different lipoproteins have different metabolisms and functional properties (Lewis, 1973). The major lipoprotein fraction linked to atherosclerosis is LDL, the dominant cholesterol ester carrier in the body (Moore & Tabas, 2011). The clearance of LDL from the circulatory system is reliant on the LDL receptor which facilitates its endocytosis into hepatic and macrophage cells. Recognition of the LDL by its receptor is based upon several specific amino acid residues located on the apolipoprotein B100 (ApoB100), the primary lipid recognition protein on LDL. These residues include lysine, arginine and histidine. Oxidative modification of LDL to oxLDL will disrupt this tightly regulated process. Oxidative induced degradation of ApoB100 results in its fragmentation forming smaller peptides and altering the positive lysine residues. This results in the failure of oxLDL to be recognised by the LDL receptor (Esterbauer, Dieber-Rotheneder, Waeg, Striegl, & Juergens, 1990; Levitan, Volkov, & Subbaiah, 2010; Obama et al., 2007). Recognition of oxLDL instead switches to macrophage scavenger receptors. Here rapid and relatively unregulated uptake can occur, resulting in the over accumulation of cholesterol within the cell and subsequent differentiation into lipid-laden foam cells (Figure 1.2) (Bobryshev, 2006; Steinbrecher, 1991). OxLDL will further facilitate extensive cellular damage triggering a cellular apoptotic or necrotic response (Katouah, Chen, Othman, & Gieseg, 2015). Cellular contents (including cellular debris, oxidants, chemotactins and cytokines) can then be released into the surrounding environment increasing inflammation. This leads to the further recruitment of macrophages and production of oxLDL, culminating in a cyclic process. (Libby, Ridker, & Hansson, 2009; Madamanchi, Vendrov, & Runge, 2005).
1.2.4 Macrophages, foam cell formation and cell death
Macrophages are immune cells derived from monocytes and are involved in both innate and adaptive immunity. Macrophage recruitment to a site of infection enables the body to trigger an appropriate response for host defence (Mosser & Edwards, 2008). This may include the removal of pathogens and other large molecules, the clearance of damaged cells and the production of other chemicals used in immune response. These benefits do not always outweigh the consequences in terms of atherosclerosis. Macrophage infiltration has been shown to play an important part in all stages of atherosclerotic plaque development from the initial thickening of the artery wall to the advanced plaque stage (Nakashima, Wight, & Sueishi, 2008). Murine models lacking monocyte-macrophage recruitment mechanisms show reduced lipid deposition into atherosclerotic lesions while histological studies indicate the presence of macrophages within lipid-rich areas of the plaque core (Potteaux et al., 2011).
As aforementioned the role of macrophages in atherosclerotic plaque development relies on their internalization of oxLDL to form lipid laden foam cells, ultimately resulting in macrophage cell death and deposition into a growing atherosclerotic plaque (Daniel Steinberg, 2009). Foam cells are characterised by their accumulation of cholesterol esters (CE) along with oxidation products similar to those seen in oxidatively modified lipoproteins (Jessup & Kritharides, 2000). Macrophages will accumulate CE and oxysterols when exposed to oxLDL (Asmis & Jelk, 2000; Boström et al., 2006; Brown, Dean, & Jessup, 1996). OxLDL has been shown to facilitate macrophage death in vitro and is suggested to be able to do this also within atherosclerotic plaques (Baird, Reid, Hampton, & Gieseg, 2005; Katouah et al., 2015). While the exact mechanisms of this process is disputed, the binding, uptake and processing of oxLDL plays a key role in the fate of a macrophage (Daniel Steinberg, 2009). When modified lipoproteins are taken up by a macrophage they are first transported to endosomes and then to lysosomes (Brown et al., 2000; Maor & Aviram, 1994). CE content is then hydrolysed into free cholesterol and fatty acids at an acidic pH. As oxLDL uptake by scavenger receptors is relatively un-regulated, its uptake leads to the accumulation of free cholesterol which can be re-esterified via ACAT leading to a large pool of esterified cholesterol within the cytosol. Experimental inhibition of ACAT also causes cholesterol accumulation in oxLDL treated cells, resulting in Endoplasmic Reticulum stress, protease activation, and apoptosis (Ozcan & Tabas, 2016; Wang et al., 1996). Under standard conditions excess cholesterol can be efluxed out of the macrophage through reverse cholesterol transport. This process is characterised by CE hydrolysis mediated by a family of enzymes with carboxyl ester hydrolase/esterase activity. Cholesterol and also cholesterol products are then efluxed out of the macrophage onto high density lipoprotein (HDL) particles (Johnson, Mahlberg, Rothblat, & Phillips, 1991) via ATP-binding cassette transporters ABCA1 and ABCG1 (Yvan-Charvet, Wang, & Tall, 2010). Yet modification of LDL has been shown to inhibit this process resulting in macrophages internalizing higher amounts of oxLDL along with reducing efflux culminating in foam cell formation.
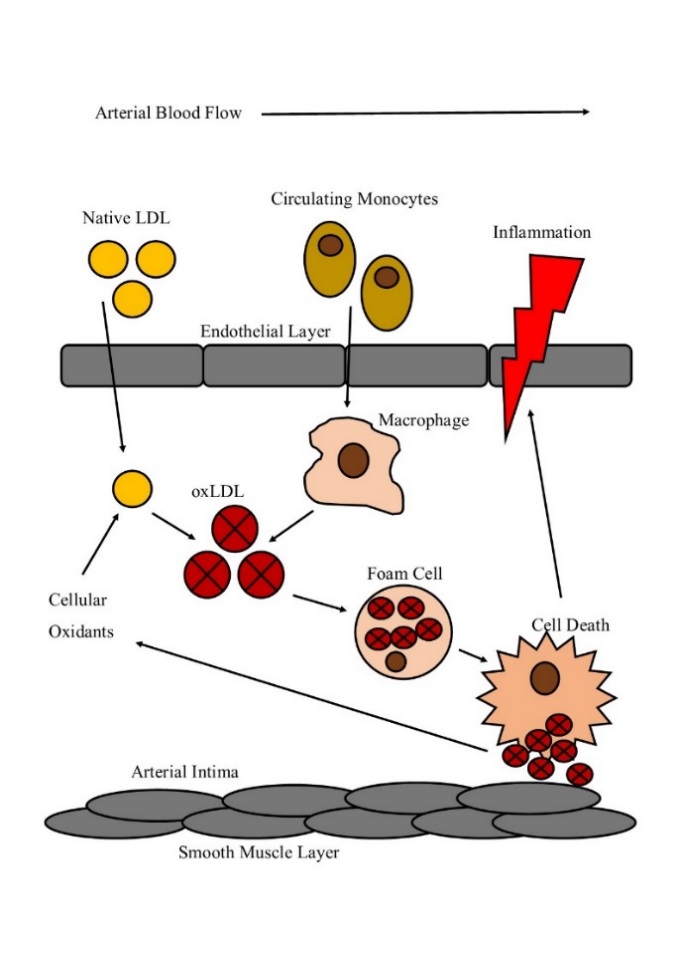
Figure 1. 2 The formation of a lipid laden foam
Native LDL enters the atrial intima space and undergoes oxidation by cellular oxidants produced by endothelial, macrophage and smooth muscle cells. OxLDL then recruits macrophage differentiated monocytes to the site of inflammation. OxLDL uptake via scavenger receptors triggers the formation of lipid loaded foam cells. Foam cell death then results in the macrophage cellular content spilling into the environment, further propagating inflammation within the intima space.
1.3 Plaque initiation theories
1.3.1 Response to injury theory
“Response to injury” is an early theory proposed by Ross (1986) to explain the initial process of plaque formation. In this model disturbance of vascular homeostasis mediated via injury is proposed to initiate atherosclerosis. Suggested triggers include disruption of laminar flow and oscillatory shear stress at the arterial bifurcation, infection of the vascular wall, along with phagocyte associated myeloperoxidase activity (Malek, Alper, & Izumo, 1999). Disturbed endothelium loses its ability to vaso-regulate making the affected area more susceptible to circulating pro-atherogenic lipoproteins (Malek et al., 1999). Endothelial injury will also recruit immune cells which will release pro-inflammatory cytokines leading to the promotion and propagation of the inflammatory response (Wick, Knoflach, & Xu, 2004). Part of this response results in the recruitment of leukocytes to the site of inflammation with the aim of resolving stress, promoting healing and can lead to the migration and adherence of monocytes to the site of endothelial inflammation. Damage caused to the endothelial layer also allows for the penetration of LDL into the arterial intima layer where oxidative modification may occur due to the inflammatory environment along with the presence of immune cells. These factors can result in foam cell formation and the growth of an atherosclerotic plaque.
1.3.2 Response to retention theory
(Williams & Tabas, 1995) suggested a second “response-to-retention” theory in which plaque development is triggered by the diffusion and retention of lipoprotein within the sub-endothelial layer of the arterial wall where it binds with high affinity to muscle associated proteoglycan chains. A major feature of proteoglycans are long carbohydrate side chains of glycosaminoglycan’s (GAGs) covalently attached to a core protein via glyosidic linkages. This theory is reliant upon the repeating disaccharide units of the GAGs which contain negatively charged sulphate or carbohydrate groups. As the LDL moves through the sub-endothelial layer the positively charged lysine groups of ApoB100 are attracted to the negatively charged sulphate proteoglycans resulting in the adherence of LDL to the GAG matrix (Evanko, Raines, Ross, Gold, & Wight, 1998; Libby et al., 2009). Retention and aggregation of lipoprotein within the vascular intima allows for its oxidative modification by oxidants formed by vascular cells and macrophages. Modification then promotes its uptake into macrophages via scavenger receptors leading to foam cell formation.
1.3.3 Oxidative modification hypothesis
The oxidative modification hypothesis is also commonly accepted in order to explain the initial stages of plaque development. This theory states that LDL particles enter the arterial intima layer where they become trapped and oxidatively modified by cellular oxidants produced either by endothelial or smooth muscle cells. These cells produce pro-inflammatory cytokines, adhesion molecules and chemotactic proteins to promote the recruitment and migration of monocytes to the inflammatory area within the intima (Hansson, 2001). Once activated monocytes can roll via cell adhesion proteins along the surface of the lumen endothelial layer triggered by a response to endothelial cell released pro-inflammatory signals. As the monocytes move through the area they undergo differentiation into macrophages in response to inflammatory cytokines. Macrophages in turn will produce oxidants such as superoxide, hydrogen peroxide and hypochlorite via the NADPH oxidase and myeloperoxidase complexes respectively, in order to oxidise any pathogenic threat. This will additionally cause further lipid and protein oxidation of LDL forming oxLDL (Bobryshev, 2006; Libby et al., 2009; Steinbrecher, 1991; Stocker & Keaney, 2004). OxLDL can then be taken up by macrophage scavenger receptors forming lipid laden foam cells which can then be added to a growing plaque (Lusis, 2000).
Inflammatory cytokines released during this process also play an important role in the destabilization of the plaques fibrous cap increasing the likelihood of plaque rupture. Released inflammatory cytokines inrerleukin-1, tumour necrosis factor, CD140 and CD-40 can stimulate an up-regulation of matrix metalloproteinases within macrophages and smooth muscle cells (Libby & Aikawa, 1998; Mach, Schönbeck, Bonnefoy, Pober, & Libby, 1997). It is these enzymes that are in part responsible for the remodelling of the extracellular matrix and the degradation of the plaques fibrous cap increasing the chances of rupture (Galis, Sukhova, Lark, & Libby, 1994; Geng et al., 1995), Once ruptured debris will form an acute thrombosis which may get lodged within the artery blocking or restricting blood supply resulting in nutrient deprivation and tissue death. This may often manifest as either a heart attack or stroke (Bobryshev, 2006; Libby et al., 2002).
1.4 Scavenger receptors
1.4.1 Scavenger receptors
As previously described the oxidative modification of LDL causing foam cell formation and cell death lead to the development of the oxidative theory of atherosclerosis. Part of the basis of this theory is the observation that cultured monocytes/macrophages will internalise oxLDL much more rapidly than unmodified LDL (Ho, Brown, Bilheimer, & Goldstein, 1976). The uptake of oxLDL is mediated via macrophage scavenger receptors. Scavenger receptors were first identified on activated macrophages and subsequently found to function in cholesterol and lipoprotein metabolism, among other roles, by binding and endocytosing a range of modified lipoproteins including oxLDL (Endemann et al., 1993). After binding, scavenger receptor-ligand interaction facilities a signalling cascade regulating macrophage activation, lipid metabolism, and inflammatory pathways (Ashraf & Gupta, 2011). Importantly in terms of atherosclerosis, and unlike the normal LDL receptor, scavenger receptors will not be down regulated by intracellular cholesterol levels allowing cholesteryl ester accumulation to the point of foam cell formation. OxLDL binding has even been shown to result in an upregulation event (D’Archivio et al., 2008). As such scavenger receptors are considered to be a major factor in the development of atherosclerotic plaques.
To date many classes of scavenger receptor have been identified and range from class A to class G. Some function as multi-ligand receptors while others only recognise specific structural motifs (Ashraf & Gupta, 2011). OxLDL is initially recognised by several scavenger receptors including the class A scavenger receptor A (SR-A) (Kodama, Reddy, Kishimoto, & Krieger, 1988), class B scavenger receptors SR-BI and CD36 (Endemann et al., 1993), class D scavenger receptor CD68 (Ramprasad, Terpstra, Kondratenko, Quehenberger, & Steinberg, 1996), and the class E scavenger receptor, lectin-like oxidised low density lipoprotein receptor-1 (LOX-1) (Yoshida, Kondratenko, Green, Steinberg, & Quehenberger, 1998). While in vivo evidence is still lacking for the particular role each plays in atherosclerosis, in vitro experimental evidence points towards an important role for CD36.
1.4.2 CD36
CD36 is an integral membrane bound multi-ligand scavenger receptor found in a number of cells including monocytes/macrophages, platelets, endothelial and smooth muscle cells (Asch, Barnwell, Silverstein, & Nachman, 1987; Collot-Teixeira, Martin, McDermott-Roe, Poston, & McGregor, 2007; Matsumoto et al., 2000; McGregor et al., 1989; Swerlick, Lee, Wick, & Lawley, 1992). CD36 present on the cell surface of macrophages plays a crucial role in the progression of atherosclerotic legions from the fatty streak to the necrotic plaque due to its ability to recognize, bind and endocytose oxLDL.
CD36 was first identified as a 88 kDa membrane glycoprotein on monocytes using the monoclonal antibody OKM5 (Talle et al., 1983). Later on it was discovered that the 88 kDa band was the incompletely glycosylated CD36 located in the Golgi apparatus, whereas the fully glycosylated 100kDa band is mature CD36 residing on the plasma membrane (Alessio et al., 1996). Further studies examining functional and structural characteristics led to CD36 being classified as a member of the class B scavenger receptor family. The gene encoding CD36 has been identified as being more than 46kb in size and located on the q11.2 band of chromosome 7 (Armesilla & Vega, 1994; Fernández-Ruiz, Armesilla, Sánchez-Madrid, & Vega, 1993). The gene consists of 15 exons, of which exons 4-13 and parts of exons 3 and 15 contribute to the protein with exons 1,2 and 15 making up the 3’ and 5’ untranslated regions.
1.4.3 CD36 structure
The structure of CD36 consists of a ditopic configuration in which an extracellular domain is flanked by two transmembrane and two cytoplasmic domains (Figure 1.3). The extra-cellular region consists of many N-linked glycosylation sites along with containing a hydrophobic domain capable of potentially interacting with the plasma membrane, a proline rich region between amino acids 242-333 and several functional domains (Greenwalt et al., 1992). One domain located between amino acids 155 to 183 and is able to bind to oxLDL, along with growth hormone releasing peptides, hexarelin, and advanced glycated products (Navazo, Daviet, Ninio, & McGregor, 1996; Ohgami et al., 2001; Pearce et al., 1998).
The binding of oxLDL to macrophage CD36 occurs via a lipid moiety (Nicholson, Frieda, Pearce, & Silverstein, 1995) differing to other scavenger receptors in which binding occurs via a apoprotein moiety. Here high affinity binding of oxLDL to CD36 appears to be related to the recognition of oxidized phospholipids covalently bound to apoB in oxLDL (Boullier et al., 2000). The main structural characteristic related here is a phospholipid with a sn-2 acyl group incorporating a terminal γ-hydroxy(or oxo)-α,β-unsaturated carbonyl which is formed during LDL oxidation (Podrez et al., 2002). A computational modelling study undertaken by Doi et al. (1993) showed that the CD36 oxLDL binding domain contains a positively charged groove made by a lysine cluster which enables specific interactions with negatively charged ligands including oxLDL. Bound oxLDL is then able to be endocytosed utilizing a raft mediated pathway seemingly separate from caveolae, caveolin-1 and clathrin mediated uptake (Zeng, Tao, Chung, Heuser, & Lublin, 2003)
Once endocytosed oxLDL brings about important transcriptional changes including the up-regulation of CD36 expression (Shiffman et al., 2000). Upon stimulation via oxLDL Peroxisome proliferator-activated receptor-γ (PPARγ) is activated via the p38 kinase pathway (M. Zhao et al., 2002) forming a heterodimer with the retinoid X receptor (RXR). This PPARγ:RXRα complex can then bind to elements in the CD36 promoter leading to increased CD36 expression (Tontonoz, Nagy, Alvarez, Thomazy, & Evans, 1998). Various kinases have also been shown to be involved in CD36 upregulation. Protein kinase C was shown to be part of oxLDL-mediated CD36 up-regulation and PPARγ activation (Feng et al., 2000) along with protein kinase B (PKB), in which PKB over expression in macrophages increased the CD36 promoter along with a PPARγ element-driven reporter gene (Munteanu et al., 2006).
STAT1 signalling also plays a role CD36 transcription. The ligand 15-Hydroxyeicosatetraenoic acid (15(S)-HETE) acts via this pathway and with inhibition of STAT1 activity significantly decreasing the expression of CD36 and subsequent foam cell formation (Agrawal et al., 2007). STAT1 activation is mediated by IFN-α, IFN-γ, and several other cytokines (Gao, 2005). IFN-γ binding to its receptor triggers a cascade of events resulting in the phosphorylation of the receptor and receptor-janus kinase proteins. Activated STAT1 can then form homodimers which when translocated to the nucleus will bind consensus DNA γ-activated sites and promote or repress various target genes including CD36 (O’Shea, Gadina, & Schreiber, 2002; Ramana, Gil, Schreiber, & Stark, 2002).
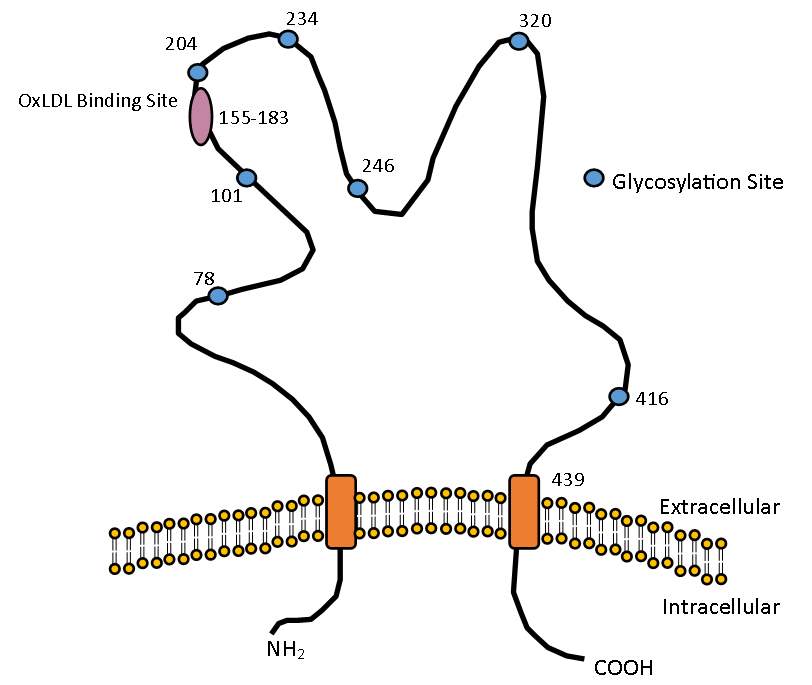
Figure 1. 3 The structure of CD36
Structure of CD36 showing the oxLDL binding site located between amino acids 155-183 and the various glycosylation sites required for maturation and transportation to the cell membrane.
1.4.4 CD36 in atherosclerosis
Initial work exploring the role of CD36 in atherosclerosis came from Endemann et al. (1993) in which the HEK293 cell line transfected with CD36 subsequently acquired the ability to specifically bind oxLDL. Later it was demonstrated that CD36, along with class A scavenger receptors, accounted for the uptake of between 75-90% of all acetylated and oxidised LDL (Kunjathoor et al., 2002). Looking at a human population Nozaki et al. demonstrated that a small subset of the Japanese population whose monocytes lack CD36 showed a 40-50% reduction in the levels of oxLDL binding to HMDMs compared to a normal population (Nozaki et al., 1995). The same levels of reduced binding were also seen when CD36 binding sites were blocked by specific monoclonal antibodies reducing oxLDL binding to HMDMS by as much as 50% (Nozaki et al., 1995), with similar results seen in a separate study usings THP-1’s (Endemann et al., 1993).
The in vivo role of CD36 is human atherosclerotic development has yet to be fully explored with most evidence coming from animal models. Mice containing a double knockout of ApoE and CD36 are a common model used. Lack of ApoE triggers very high circulating levels of cholesterol rich lipoproteins and enables the development of atherosclerotic lesions at a much faster rate than wild type populations. After 12 weeks ApoE/CD36 knockout mice exhibit a 76.5% decrease in aortic lesion size compared to mice only lacking AopE, while 60% less oxLDL was internalized in the double knockouts compared to ApoE alone (Febbraio et al., 2000). Further evidence for role of CD36 in plaque development came when CD36 was reintroduced to knockout mice via a stem-cell transfer, resulting in an increased size of the atherosclerotic lesion area (Febbraio et al., 2000). The sum total of these and similar publications demonstrates the key role that this scavenger receptor plays via oxLDL uptake and foam cell formation in atherosclerotic development.
1.5 7,8-Dihydroneopterin
1.5.1 GTP, 7,8-Dihydydroneopterin and products
Interferon-γ (INF-γ) is a dimerized & soluble cytokine critical in both innate and adaptive immunity. INF-γ has been reported to be involved in the regulation of over 500 genes (Leon & Zuckerman, 2005) and has been associated with both pro- and anti-atherogenic properties. One of INF-γ’s role in atherosclerosis relates to the stimulation of the production of 7,8-diydrneopterin and neopterin (Huber et al., 1984) which have both been detected within atherosclerotic plaques and associated with cardiovascular events (Janmale et al., 2015). Neopterin is considered to be a viable marker of plaque stability and atherosclerotic prognosis (Liu & Li, 2013), while 7,8-NP has been demonstrated in vitro to have a protective role against oxLDL mediated cell death (Steven Gieseg, Leake, et al., 2008). 7,8-NP and neopterin belong to the pteridine class of compounds which as a common characteristic have a pyrazino-2,3-pyrimidine bicyclic nitrogen containing ring system, and depending on the size of substituent groups can further be described and conjugated or un-conjugated (Wachter et al., 1992). Further sub-classification for unconjugated pteridines is determined on the corresponding oxidation state composing of the fully reduced tetrahydropterins, to the partially reduced dihydroneopterins and aromatic pterins (Oettl & Reibnegger, 2002).
All members of the pteridine class of compounds are synthesized from a guanosine triphosphate (GTP) precursor (Figure 1.4). INF-γ stimulates the first step via the enzyme GTP cyclohydrolase 1 which metabolises GTP into 7,8-NP triphosphate (Werner et al., 1990). Phosphatase enzymes them hydrolyse 7,8-NP triphosphate to generate 7,8-NP (Wachter et al., 1992). Co-stimulation with tumour necrosis factor-α (TNFα), dexamethasone or LPS has also been demonstrated to enhance synthesis (Werner-Felmayer et al., 1995; Werner-Felmayer et al., 1990) although not as strongly as INF-γ. Breakdown of 7,8-NP after reaction with oxidant species will form neopterin, 7,8-dihydroxanthopterin and xanthopterin.
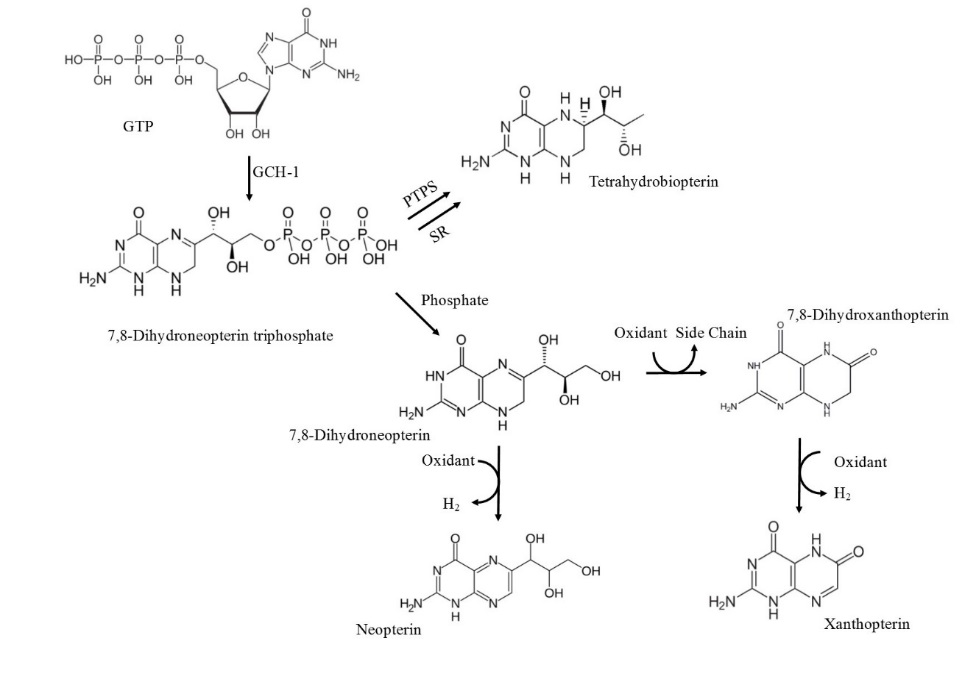
Figure 1. 4 The biosynthetic pathway of 7,8-NP and its reduced products
The biosynthetic pathway of 7,8-NP involves the conversion of GTP to 7,8-dihydroneopterin triphosphate (7,8-DNT) catalysed by GTP cyclohydrolase-I (GCH-I). Accumulation of 7,8-DNT in cells of a myelocytic origin drives the dephosphorisation of 7,8-DNT to 7,8-NO due to insufficient 6-pyruvonyltetrahydropterin synthase (PTPS). Alternative cells with functional PTPS will drive tetrahydrobiopterin formation. Breakdown of 7,8-NP after reaction with oxidant species will form neopterin, 7,8-dihydroxanthopterin and xanthopterin. Adapted (Dántola, Vignoni, Capparelli, Lorente, & Thomas, 2008; Fuchs et al., 2009)
1.5.2 7,8-NP as an antioxidant
The atherosclerotic plaque is a site of chronic inflammation as indicated by the high number of immune cells such as macrophages, and the presence of various inflammatory markers. Macrophage cells can release a range of oxidizing agents including superoxide, hydrogen peroxide, lipid peroxides, lipoxygenases and possibly hypochlorite, which are all known to contribute to the production of oxLDL (Steven Gieseg, Leake, et al., 2008). While the oxidative theory of atherosclerosis has come under some criticism due to the inability of various antioxidant intervention trials to exhibit protection, these have focused on ascorbate and tocopherol which are tightly controlled in vivo and at high levels can act as pro-oxidants. Reduced pterins, such as 7,8-NP, have also been shown to act as antioxidants and are produced by macrophage cells in the atherosclerotic plaque. 7,8-NP has been demonstrated to have a protective effect with in vitro studies showing its ability to interfere with LDL oxidation (Steven Gieseg et al., 1995), reactive oxygen species (ROS) mediated reactions (Oettl, Greilberger, Dikalov, & Reibnegger, 2004) and free radical damage including from hydroxyl and peroxyl radicals (Duggan, Rait, Platt, & Gieseg, 2002)
7,8-NP is a scavenger of superoxide and peroxyl radicals generated via 2,2’-azobis-2-methyl-propanimidamide dihydrochloride (AAPH) (Oettl et al., 2004) while also reacting with hydrogen peroxide and chloramine-T (Weiss et al., 1993). The reaction rate of 7,8-NP with AAPH derived peroxyl radicals which nears the rate of diffusion makes it a very good inhibitor of this oxidation pathway (Duggan, Rait, Gebicki, & Gieseg, 2001). 7,8-NP has also been shown to inhibit oxidation of the polyunsaturated omega-6 fatty acid linoleate via AAPH and the formation of diene on LDL particles during both AAPH- and copper-mediated oxidation of LDL (Steven Gieseg et al., 1995). Another biologically relevant experimental system showed a 7,8-NP antioxidant effect in protecting bovine serum albumin from AAPH–mediated oxidation (Duggan et al., 2001).
7,8-NP causes a reduction in erythrocyte haemolysis induced by HOCl, H2O2 and AAPH treatments (Steven Gieseg, Maghzal, & Glubb, 2001; Y.-t. T. Yang, Whiteman, & Gieseg, 2012). Complete protection was observed in the presence of AAPH and partial protection (40%) in the presence of H2O2 (Steven Gieseg et al., 2001). 7,8-NP has also been reported to protect against lipid hydroperoxide formation and protein oxidation (Steven Gieseg et al., 2001). Additional studies confirmed this effect of 7,8-NP in which protein hydroxide and lipid peroxide formation on LDL was slowed in HMDM and THP-1 cells (Firth, Crone, Flavall, Roake, & Gieseg, 2008).
Further studies into protective roles of 7,8-NP have shown its ability to protects cells of myelocytic origin from the cytotoxic effects of oxLDL. It has been found that in monocyte like THP-1 and HMDM cells oxLDL formation was fully inhibited with only micro molar concentrations of 7,8-NP (Steven Gieseg & Cato, 2003), while a separate study showed that 7,8-NP protected U937 cells from the cytotoxicity of oxLDL (Baird et al., 2005). This protective role is thought to occur via the use of 7,8-NP’s antioxidant properties to scavenge any ROS that may be generated in response to oxLDL.
1.5.3 Antioxidant down regulation of CD36
Besides 7,8-NP’s antioxidant properties a second mode of protection is hypothesised to occur via the down regulation of CD36 itself, preventing oxLDL uptake and foam cell formation (Figure 1.5). In HMDM cells the addition of 7,8-NP triggered the down regulation of the 100-kDa form of CD36 and partial reduction of the immature intracellular 88-kDA form, while also reducing the levels of reactive oxygen species (S. Gieseg et al., 2010). 7,8-NP is not the only antioxidant known to down regulate CD36 expression. α-tocopherol (Vitamin E) is well known as a lipophilic antioxidant and has been shown to inhibit oxLDL uptake by the down regulation of CD36 mRNA and protein expression (Ricciarelli, Zingg, & Azzi, 2000). Yet the same effect was not seen when cells are given β-tocopherol or probucol thus ruling out a general anti-oxidant regulated mechanism of down regulation (Ricciarelli et al., 2000). One alternative mechanism is that α-tocopherol inhibited protein kinase C (PKC) activity and it is the phosphorylation/dephosphorylation of the CD36 PPARγ transcription factor that may be involved in the down regulation of CD36. A second proposed mechanism was that α-tocopherol could modulate gene expression indirectly via inhibiting the activated oxidised form of PPARγ ligands. Evidence of α-tocopherol acting by this pathway came from an experiment showing that it reduced the level of PPARγ activators 9-Hydroxyoctadecadienoic acid (9-HODE) and 13-HODE by inhibiting their enzymatic production (Belkner, Stender, & Kühn, 1998).
1.5.4 Other modes of CD36 down regulation
Transforming growth factor-β1 (TGF-β1) and TGF-β2 have been shown to down- regulate CD36. Han et al. showed that TGF-β1 and TGF-β2 decreased expression of CD36 in THP-1 cells by a mechanism involving phosphorylation of mitogen activated protein (MAP) kinase which in turn initiated phosphorylation of PPARγ leading to a decrease in CD36 gene transcription (J. Han et al., 2000).
Alongside the transforming growth factors Boyer et al. showed a mechanism for CD36 down regulation in the form of the cytokine TNF-α which is known to play a major role in the development of atherosclerosis (Boyer et al., 2007). Using human monocytes Boyer et al. showed that TNF-α inhibited both CD36 membrane expression and mRNA expression in a process involving a reduction in PPARγ activation (Boyer et al., 2007). These results corresponded well to studies in which a reduction in PPARγ activation by TNF-α was reported in human adipocytes and hepatocytes (Sung, She, Xiong, & Tsukamoto, 2004). Further evidence for the role that this cytokine plays in CD36 down regulation came from a study by Zamora et al. who showed TNF-α trigged CD36 down regulation in toll like receptor (TLR) 2 (Pam3CSK4 and FSL1) and TLR4 ligand lipopolysaccharide (LPS) stimulated human peripheral blood mononuclear cells (PMBC) (Zamora et al., 2012). Additional anti-inflammatory cytokines have also been suggested to play a role in CD36 down regulation,. Rubic et al. using THP-1 cells and peripheral monocytes showed that anti-inflammatory cytokine interleukin-10 (IL-10) suppressed basal and PPARγ stimulated transcription of CD36 due to reduced PPARγ protein expression (Rubic & Lorenz, 2006). However this is contrasted in the study by Zamora et al. (2012) who although agreed that IL-10 along with IL-6 were produced during TLR induced down regulation, they themselves were not able to explain this effect.
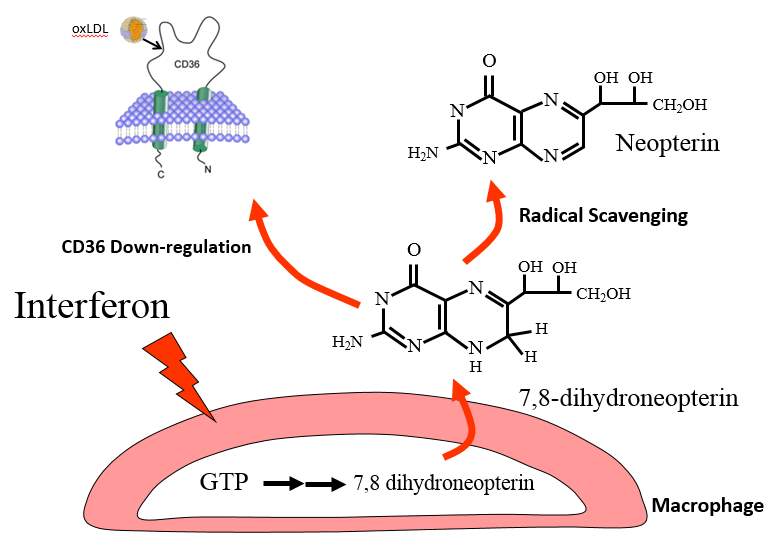
Figure 1. 5 Protective mechanisms of 7,8-NP
7,8-NP produced by GTP stimulated macrophages is thought to protect against atherosclerosis by scavenging radicals and in so preventing oxLDL and subsequent foam cell formation, along with down-regulating CD36, the main scavenger receptor of oxLDL.
1.6 Peroxisome proliferator activated receptor-ɣ
CD36 transcription is known to be controlled by the heterodimer Peroxisome Proliferator Activated Receptor-γ/Retinoid X Receptor (PPARγ/RXR) (S. Han & Sidell, 2002) with PPARγ ligands 15-deoxyD12 and 14-prostaglandin J2 increasing CD36 expression (Feng et al., 2000). PPARγ is a member of the PPAR subfamily of nuclear hormone receptors which act as ligand-activated transcription factor, regulating gene expression by interacting with specific DNA sequences upstream of its target genes. The PPAR subfamily names relates to the ability for the original member PPARα, to induce peroxisome proliferation in response to a variety of compounds (S. Lee et al., 1995). Other PPAR members do not necessarily share this function, instead acting as major regulators of the numerous aspects of lipid metabolism and metabolic control. Using PPARα as a base PPARγ was first identified using homology cloning in Xenopus (Dreyer et al., 1992) and subsequently in mice (Zhu, Alvares, Huang, Rao, & Reddy, 1993). Afterwards it was discovered to act as the main regulator of adipogenic differentiation (Tontonoz & Spiegelman, 2008).
1.6.1 PPARγ structure and function
Common to all members of the PPAR subfamily, PPARγ forms an obligate heterodimer with RXR in order to carry out its function (Kliewer et al. 1997), with lack of dimerization compromising PPARγ’s ability to bind to DNA with high affinity. The C-terminal region of PPARγ is required for RXR dimerization and also contains AF2, the major transcriptional activation domain, along with forming a ligand-binding pocket (Tontonoz 2008). The 120 amino acids that make up the N-terminal region exhibits transcriptional activity when interacting with a heterologous DNA-binding domain. Contradictory evidence using deletion studies have shown that when the N-terminal domain is removed PPARγ exhibits increased greater transcriptional activity (Tontonoz, Hu, & Spiegelman, 1994) suggesting that this domain may also contribute a level of inhibitory function.
1.6.2 Modulation of PPARγ activity
PPARγ is a phosphoprotein in which it can be post-translationally modified by the attachment of a phosphate group by MAP Kinases (Figure 1.6). In contrast to the likes of insulin this modification decreases transcriptional activity (Adams, Reginato, Shao, Lazar, & Chatterjee, 1997; Hu, Kim, Sarraf, & Spiegelman, 1996). In vitro assays have shown that two MAP Kinases ERK2, and JNK are able to phosphorylate the PPARγ leading to a decrease in transcriptional activity, including that of CD36 (Adams et al., 1997; Camp & Tafuri, 1997). Phosphorylation mediate by both these MAP kinases has been shown to occur at the same site at serine 82 of mouse PPARγ1 (Shao et al., 1998). The substitution by alanine here causes the loss of platelet-derived growth factor mediated repression of PPARγ activity (Camp & Tafuri, 1997). In humans phosphorylation at the corresponding S84 site was shown to inhibit ligand dependent as well and independent trans-activating function. A separate study also showed that mutation at the MAP kinase binding site lead to a decrease in ligand binding affinity (J. Han et al., 2000) which may suggest communication between phosphorylation and ligand binding sites, similar to that seen in PPARα. In murine models PPARγ 2 is able to be phosphorylated at serine 112 culminating in decreased PPARγ activity (Adams et al., 1997; Hu et al., 1996). In macrophages TGF-β increased PPARγ phosphorylation and subsequently decreased thiazolidinedione (TZD) (a PPAR activator) induced CD36 expression (S. Han & Sidell, 2002).
PPARγ function can also be regulated by ligand binding. Bound ligands induce a conformational change which allows for the release of transcriptional inhibitors and the recruitment of transcriptional co-activators. One class of ligands involves redox-regulation. The oxidised lipid 15-deoxy-Δ12,14-PGJ2 has been shown to act as a ligand (Forman et al., 1995), along with 9-(HODE), 13-(HODE) (Lenz et al., 1990), and 15(S)-HETE (Nagy, Tontonoz, Alvarez, Chen, & Evans, 1998).
One pathway of ligand activation involves the STAT1 signalling. Activated STAT1 forms a homodimer which when translocated to the nucleus will bind consensus DNA γ-activated sites and activate transcription (O’Shea, Gadina, & Schreiber, 2002; Ramana, Gil, Schreiber, & Stark, 2002). Interaction of STAT1 with PPARγ requires it to become actytelated, but not phosphorylated. Acetylation requires p300 activation which in turn requires Syk2 and Pyk2 stimulation and XO and NADPH oxidise mediated ROS production. Although the exact mechanism of how 15(S)-HETE activates XO/NADPH activity is not yet known.
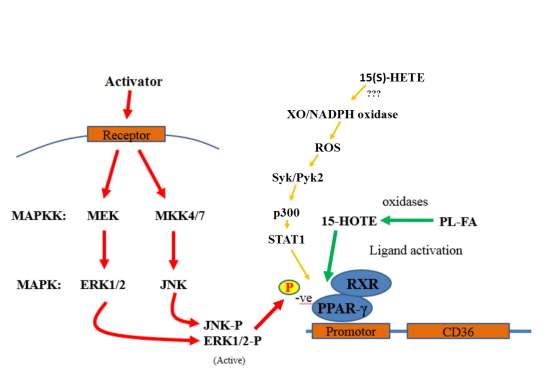
Figure 1. 6 Regulation mechanics of PPARγ
The regulation mechanics of PPARγ induced down regulation shows a activated MAP Kinase cascade within the macrophage resulting in the phosphorylation of PPARγ’s kinase binding site subsequently decreasing the transcriptional activity for CD36. Oxidised ligand and STAT1 activation of PPARγ is also shown.
1.7. MAP kinases
Mitogen-activated protein kinases (MAP kinase) are a highly conserved family of protein kinases specific to the amino acids serine, threonine, and tyrosine. MAP kinase signalling pathways are involved in a wide variety of cellular functions including proliferation, differentiation, motility, stress response, apoptosis, and survival. Classic MAP kinases include the extracellular signal-regulated kinase 1 and 2 (Erk1/2), the c-Jun N-terminal kinases 1-3 (JNK1-3)/ the p38 isoforms (p38α, β, γ, and δ) and Erk5. These share number of characteristics including similar substrate sites, phosphorylation dependent activation and a “three-tiered” pathway structure in which phosphorylation cascades transmit movement of the signal from one “tier” to the next.
A diverse range of extracellular stimuli trigger MAP Kinase activation. Such stimuli include mitogens, cytokines and growth factors. A stimuli will initially activate one or more MAP kinase kinase kinases (MAPKKKs) either by a receptor dependent or independent mechanism in the initial “tier”. MAPKKKs can then phosphorylate and activate a downstream MAP kinase kinase (MAPKK), the second “tier” which in turn phosphorylates and activates MAP kinase. MAP kinase activation can then trigger the phosphorylation and activation of MAP kinase-activated protein kinases (MAPKAPKs) which function to amplify and mediate the wide range of biological process required.
1.7.1 ERK cascade
ERK MAP kinases (otherwise known as the classical MAP kinase) cascades are involved in a wide variety of cellular functions including in adhesion, progression, migration, survival, differentiation, metabolism, proliferation, and transcription (Roskoski, 2012). ERK1/2 is activated by dual phosphorylation on tyrosine and threonine by the MAPKK MEK1 and MEK2 (Cobb, Boulton, & Robbins, 1991). These in turn are activated by the MAPKKK’s Raf-1, B-af and c-MOS. Termination of ERK1/2 activation is dependent on regulatory mechanisms that remove one ore both phosphates by tyrosine or serine/threonine phosphatases (Todd, Tanner, & Denu, 1999).
1.7.2 JNK cascade
JNK kinase cascades are involved in mediating the cellular apoptotic response to pro-inflammatory genotoxins along with having a role in the regulation of cell proliferation, survival and differentiation (C. Davies & Tournier, 2012). JNK is selectively phosphorylated and activated by JNKK’s MKK4 and SEK, which in turn are activated by the MAPKKK’s MEKK1/2 and Tpl-2.
1.7.3 P38 cascade
The p38 MAP kinase and its signalling cascades are activated by stress and plays a role in immune response, cellular survival and differentiation (Cuadrado & Nebreda, 2010). Four p38 MAP kinase isoforms have been identified with the p38α and p38β isoforms being ubiquitously expressed (Ono & Han, 2000). The p38 pathway is activated by the MAPKK’s MKK3, JNKK and MEK6 and MAPKKK’s Rac-1, Cdc42 and PAK. In most inflammatory cells p38α is the predominately activated isoform with extracellular stimuli coming from a variety of cytokines and a number of pathogens that activate though different toll-like receptors. Several growth factors as well as environmental factors such as heat shock, osmotic regulation, UV light, hypoxic stress and oxygen radicals and also capable of inducing activation.
1.7.4 MAP kinases & atherosclerosis
Reactive oxygen species such as superoxide and hydrogen peroxide have been shown to be produced in vascular smooth muscle cells, endothelial cells and cardiomyocytes (Abe & Berk, 1998; Griendling, Sorescu, & Ushio-Fukai, 2000; Yoshizumi, Tsuchiya, & Tamaki, 2001). There is evidence that MAP kinases play a role in vascular remodelling alongside these reactive oxygen species. MAP kinases have been shown to be impacted by oxidative stress with hydrogen peroxide significantly stimulating ERK1/2, JNK and p38 is rat aortic smooth muscle cells (SMC) (Yoshizumi, Abe, Haendeler, Huang, & Berk, 2000) Angiotensin II and endotheilan I also activate MAP kinases in VSMC and have been suggested to be a cause of vascular remodelling (Ishida, Ishida, Thomas, & Berk, 1998) (Yoshizumi et al., 1998).
1.8 Objective of research
OxLDL uptake by macrophage CD36 leading to foam cell formation is one process at the core of atherosclerotic plaque development. Mechanisms that are able to decrease levels of CD36 are of increasing interest in order to slow the progression of atherosclerosis. One potential mechanism for CD36 down regulation involves the human macrophage derived anti-oxidant 7,8-NP which has been shown to decrease basal CD36 levels in HMDM. In this study it is hypothesised the 7,8-NP is exerting its effect via interaction with the PPARγ transcription factor. Specifically it is thought that 7,8-NP is activating a MAP Kinase cascade with the end result being the phosphorylation of the PPARγ MAP kinase binding site and a subsequent decrease in CD36 transcription.
The first objective of this research will be to confirm that the 7,8-NP induced down-regulation seen in HMDM occurs in the U937 monocyte like cell line. This will include looking at the effects of time and concentration on CD36 levels, and also whether recovery after down regulation is possible. 7,8-NP induced down-regulation will be explored mainly using western blotting with cell lysate after experimental treatment being immunoblotted for the CD36 antibody. The effectiveness of flow-cytometry will additionally be explored using CD36 antibody binding to the cell surface of whole cells. The next part of this thesis will confirm that CD36 transcription is under the control of PPARγ in the U937 cell line using a selective PPARγ inhibitor in order to confirm our choice of target. It will also be investigated whether 7,8-NP alternatively has its effect via enhanced CD36 degradation using the protein synthesis inhibitor cycloheximide. The research will then explore the effects of MAP Kinases on 7,8-NP induced CD36 down-regulation. This will be achieved using selective MAP Kinase inhibitors including those to ERK1/2, JNK and p38. The effect of NF-κB inhibition will also be explored.
This research will help determined the mechanism via which 7,8-NP induces CD36 down regulation and help to improve our understanding on the role the 7,8-NP plays in the development of atherosclerosis.
2. MATERIALS & METHODS
2.1 Reagents, media and buffers
All reagents were of analytical grade. All solutions were prepared with deionized water purified with a Milli-Q ultrafiltration system (Millipore, Massachusetts, USA). This water is referred to as Nano-pure water in this thesis.
2.1.1 Reagents
β-Mercaptoethanol Sigma Chemical Co., Missouri, USA
4-Morpholine-propanesulfonic acid (MOPS) Sigma Chemical Co., Missouri, USA
7,8-Dihydroneopterin (7,8-NP) Schircks Laboratory, Switzerland
Acetic acid (glacial) BDH Lab Supplies, Poole, England
Anchor non-fat milk powder Fonterra Ltd, New Zealand
BAY 11-7082 (NF-κB Inhibitor) Sigma-Aldrich Co. LLC, New Zealand
Bicinchoninic acid (BCA) protein determination kit Pierce Biotechnolgy Inc., Illionois, USA
Bovine serum albumin (BSA) Sigma-Aldrich Co. LLC, New Zealand
Bromophenol blue Sigma-Aldrich Co. LLC, New Zealand
cOmplete™, Mini Protease Inhibitor Cocktail Sigma-Aldrich Co. LLC, New Zealand
Coumassie blue Bio-Rad Laboratories, California, USA
Dimethyl sufloxide (DMSO) BDH Lab Supplies Ltd, Poole, England
Ethanol BDH Lab Supplies Ltd, Poole, England
Ethylene-diamine-tetra-acetic acid (EDTA) Sigma-Aldrich Co. LLC, New Zealand
Glycerol Sigma-Aldrich Co. LLC, New Zealand
GW9662 (PPAR-ɣ inhibitor) Sigma-Aldrich Co. LLC, New Zealand
Hydrochloric acid (HCl) Merck, Germnay
Methanol Merck, Fischer Scientific, Belgium
Molecular weight marker Bio-Rad Laboratories, California, USA
Neopterin Schircks Laboratory, Switzerland
NuPAGE 4-12% Bis-tris Gels, 1.0mm x10 well Invitrogen, California, USA
NuPAGE 4-12% Bis-tris Gels, 1.5mm x15 well Invitrogen, California, USA
PD98059 (MEK Inhibitor) Sigma-Aldrich Co. LLC, New Zealand
Phorbol 12-myristate 13-acetate (PMA) Sigma Chemical Co., Missouri, USA
Ponceau S Sigma Chemical Co., Missouri, USA
SB202190 (p38 inhibitor) Sigma-Aldrich Co. LLC, New Zealand
Sodium bicarbonate (NaHCO3) Sigma-Aldrich Co. LLC, New Zealand
Sodium chloride (NaCl) BDH Lab Supplies Ltd, Poole, England
Sodium dodecyl sulphate (SDS) Sigma-Aldrich Co. LLC, New Zealand
Sodium hydroxide (NaOH) BDH Lab Supplies Ltd, Poole, England
SP600125 (JNK Inhibitor) Sigma-Aldrich Co. LLC, New Zealand
Supersignal West Dura Chemiluminescence Pierce Biotechnolgy Inc., Illionois, USA
Thimerosal Sigma Chemical Co., Missouri, USA
Tris(hydroxymethyl)aminomethane Sigma-Aldrich Co. LLC, New Zealand
Triton X100 Sigma-Aldrich Co. LLC, New Zealand
Trypan blue solution (0.4%) Sigma-Aldrich Co. LLC, New Zealand
Tween 20® Sigma-Aldrich Co. LLC, New Zealand
2.1.2 Media
Penicillin/Streptomycin solution: Invitrogen, Life Technologies, New Zealand 1000 U of Penicillin G & 1000 µg of Streptomycin/mL
RPMI 1640 media, with phenol red Sigma-Aldrich Co. LLC, New Zealand
RPMI 1640 media, without phenol red Sigma-Aldrich Co. LLC, New Zealand
2.1.3 General solutions and buffers
1. Roswell Park Memorial Institute (RPMI)-1640 media (with or without phenol red)
Powered RPMI-1640 (Sigma-Aldrich Co. LLC, New Zealand) was prepared in accordance to the manufacturer’s instructions. RPMI-1640 was dissolved in nano-pure water before addition of sodium bicarbonate (NaHCO3) and adjustment of pH to 7.4 by the addition of 11.4M hydrochloric acid (HCl). The RPMI-1640 medium was next sterilized by filtration through a 0.22 µm Millex® filter (Sartorius AG, Goettingen, Germany) and a peristaltic pump (CP-600, Life Technologies, Maryland, USA) into sterile 500 mL bottles previously washed with nitric acid and autoclaved. Prepared media was stored at 4oC.
Premade sterilized RPMI-1640 was also used (HycloneTM, GE Healthcare Life Sciences, Utah, USA) pre-supplemented with sodium bicarbonate, pH 7.4
2. Phosphate buffered saline (PBS)
PBS buffer consisted of 150mM sodium chloride (NaCl) and 10mM sodium dihydrogen orthophosphate (NaH2PO4) at pH 7.4. The solution was prepared by mixing 50 mL of 3M NaCl, 40 mL of 250mM NaH2PO4, pH 7.4 and 910 mLof nano-pure water. PBS required for sterile cell culture experiments was sterilized by autoclaving (15 minutes, 121oC, 15 PSI).
3. 7,8-dihydroneopterin solution
A 1mM 7,8-Dihydroneopterin (7,8-NP) (Schircks Laboratories, Switzerland) stock solution was prepared in RPMI 1640 media, in a 15 mL screw top centrifuge tube (Greiner, Greiner Bio-one, Neuburg, Germany). The solution was sonicated for 10 minutes or until dissolved before being sterilized by passage through a 0.22µM PES syringe filter (Membrane Solution, USA). The 7,8-NP solution was kept on ice at all times and used immediately after preparation.
4. D-neopterin solution
Neopterin stock solution (1mM) was prepared by dissolving D-neopterin powder (Schircks Laboratories, Switzerland) in RPMI-1640 via sonication in a 15 mL screw top centrifuge tube (Greiner, Greiner Bio-one, Neuburg, Germany). The solution was sonicated until dissolved before being sterilized by passage through a 0.22µM PES syringe filter (Membrane Solution, USA). The neopterin solution was kept on ice at all times and used immediately after preparation.
5. Triton lysis solution
Lysis buffer consisted of 0.5% Triton-X100 (diluted from 1% stock solution in nano-pure water). Protease inhibitor (ProteaseMini, Roche Diagnostics) prepared and frozen to the manufacturers recommendations (ProteaseMini, Roche Diagnostics), was defrosted and added to the lysis solution on the day of use.
6. MOPS buffer for SDS-PAGE
A 10x stock solution of MOPS was made with 500mM MOPS, 500mM Tris base, 1% SDS (w/v), and 10mM EDTA in water with pH adjusted to 7.7 (adjusted with concentrated HCl). Stock solution was diluted with nano-pure water to 1x concentration before use.
7. Transfer buffer for western blotting
Transfer buffer consisted of 25mM Tris, 200mM glycine and 20% methanol (v/v) in water and was stored at 4oC. Transfer buffer was able to be reused once.
8. Tris-buffered saline (TBS)
TBS used for nitrocellulose membrane washing, consisted of 40mM Tris-HCl, 150mM NaCl, 0.05% Tween20 (w/v) and 0.01% thimerosal (w/v) dissolved in nano-pure water. Final pH was adjusted to 7.5 using concentrated HCl.
9. TBSM
The membrane blocking solution (TBS-milk, TBSM) consisted of 5% (w/v) of nonfat milk powder (Anchor, New Zealand) dissolved in TBS. It was stored at 4◦C for up to one week.
10. Cracker buffer for SDS-PAGE
To make the cracker buffer, 125mM Tris-HCl, pH 6.8 (adjusted with concentrated HCl), 1% SDS (w/v), 20% glycerol (w/v) and 0.1% bromophenol blue (w/v) were mixed in nano-pure water. Immediately prior to use, 2% (v/v) of β–mercaptoethanol and 0.5mM EDTA was added to the amount required of the above solution.
11. Restore stripping buffer
Restore stripping buffer solution used to remove antibodies from the nitrocellulose membrane in preparation for reprobing consisted of 2 % SDS, 50mM Tris pH 6.8 , and 100µM β–mercaptoethanol and made up to volume in nano-pure water.
12. Ponceau S stain
Ponceau S stain consisted of 0.01% Ponceau S (w/v) in 5% acetic acid (v/v).
13. Propidium iodide solution
A 1 mg/mL stock solution of propidium iodide dye (Sigma-Aldrich Co. LLC, New Zealand) was prepared in nano-pure water and was kept in a sterilized schott bottle, wrapped in tin foil at 4oC
14. Phorbol 12-myristate 12-acetate (PMA)
A 5mM PMA stock solution was prepared by dissolving PMA powder in DMSO. 20 µL aliquots were pipetted into 1.7 mL centrifuge tubes and stored at -20oC
2.2 Methods
2.2.1 Cell culture based experiments
2.2.1.1 Aseptic technique
All work involving cell culture was performed under aseptic conditions using a Class II biological safety cabinet (Clyde-Apex BH 200). All cell culture equipment used was sterile-brought plastic ware (Falcon, Terumo, Unomedical, Greiner Bio-one) or had been sterilized by autoclaving (15 miniutes, 121oC, 15 PSI). All media/solutions added to cells was sterilized by autoclaving or by filtration using a 0.22 µm filter (Membrane Solutions, USA). All equipment was sprayed with 70% ethanol (diluted from stock solution in distilled water) before use in the Class II safety cabinet. Cells were kept incubated in a humidified atmosphere at 37°C containing 95% air w/ 5% CO2 (Sunnyo Electric Cp. LTD, Japan).
2.2.1.2 Standard RPMI-1640 media for U937 cell culture
RPMI-1640 with phenol red enriched with 100 U/mL penicillin G, 100 µg/mL streptomycin and 5% (v/v) foetal bovine serum (FBS) was used as standard in U937 cell culture maintenance.
2.2.1.3 Preparation and culture of the U937 cell line
The U937 cell line is a type of immortal cell line derived from the malignant cells of a pleural effusion from a patient with diffuse histiocytic lymphoma, and are one of the few human cell lines expressing monocyte like characteristics exhibited by cells of histiocytic origin. The U937 cells used in this work were from a line gifted by the Haematology Resesearch Laboratory of the Christchurch School of Medicine, University of Otago.
Prior to revival stock U937 cells were stored in liquid nitrogen using 1 mL storage vials containing 10×106 cells/mL in dimethyl sulfoxide (DMSO) freezing medium. To prepare stock U937 for use one vial was defrosted in a 37oC water bath until almost fully thawed. This suspension was next poured into 20 mL of RPMI-1640 and centrifuged at 500g for 5 minutes in order to separate the DMSO from the cells. The cell pellet was re-suspended in 10 mL of RPMI-1640 before being cultured in 25 cm3 tissue culture flasks (Flacon, BD, USA) until the cells were deemed fully recovered from their storage state and transferred into a standard 75 cm3 tissue culture flask (Greiner Bio-one, Germany).
Cells were deemed suitable for experimental work once they had reached an approximate density of ~1×106 cells/mL and division rate had normalized to a doubling time of 92-96 hours. These cells showed a spherical morphology with small grain like protrusions on the membrane from cytoplasmic organelles, and with little clustering or blebbing. Subsequent cell density was maintained between 0.3-1.5×106 cells/mL by passaging every 2-3 days.
2.2.1.4 Cell culture experimental conditions and procedures
Experiments with the U937 cell line were performed using either 12 or 24-well suspension culture plates (Cellstar ®, Greiner Bio-one). The total volume used during experimental work was 1 mL per well. Cells were counted using a haemocytometer in conjunction with a light microscope after dilution with trypan blue (ratio of 1:1). The required amount of cells was centrifuged at 500 g for 5 minutes at room temperature then re-suspended in the required experimental medium pre-heated to 37oC using a water bath. The cells were then aliquoted into the plate wells to a final concentration of 0.5×106 cells/mL.
2.2.2 Cell viability assays
2.2.2.1 Propidium iodide cell viability assay
U937 cell viability was measured after experimental treatment via the use of propidium iodide (PI) staining and flow cytometery (BD AccuriTM C6 flow cytometer, BD Biosciences, California, USA). PI is able to intercalate between DNA bases when the cell membrane becomes compromised during cell death. Intercalation can be measured via fluorescence at an excitation wavelength of 535 nM and emission wavelength of 617 nM. Combined with the use of a flow cytometer PI can be used to examine cell viability along with identifying necrotic, apoptotic and healthy cells.
After experimental treatment 250 µL of cell suspension was incubated with 4 µL of 1mg/mL PI and mixed via inversion. Incubation was allowed to occur in the dark for 10 minutes. Viability was measured via using a set volume (30 µL) analyzed on the flow cytometer using forward scatter (FSC) and the fluorescence filter FL-3 (FL-3). Cellular debris were identified and excluded from analysis and the cellular viability ratio (PI+ve:PI-ve) was compared to control samples.
2.2.2.2 Trypan blue viability test
The trypan blue viability tests is a dye exclusion test which is used to determine the number of viable cells present in a cell suspension. Live cells contain intact cell membranes that exclude the Trypan Blue dye, whereas non-viable cells have compromised cellular membranes that allow for the dye to enter the cellular cytoplasm staining it blue.
After experimental treatment the cellular suspension was diluted in a 1:1 ratio with 0.4% Trypan Blue solution and incubated for two minutes. The suspension was then transferred to a hemocytometer and the number of viable and non-viable cells counted. Viable cells were identified as having a white or clear cytoplasm, while non-viable cells were identified as having a blue cytoplasm. The ratio of viable:non-viable cells was then calculated and cellular viability determined.
2.2.3 Determination of protein concentration
Protein concentration of cell lysate was quantified using the bicinchoninic acid (BCA) protein determination kit assay (PierceTM, Rockford, USA) in which the reduction of Cu2+ to Cu+ by protein in an alkaline medium can be measured via a BCA reagent. The reaction product of this assay exhibits strong absorbance at 562 nM with increasing protein concentration in the range of 25–250µg/mL.
Fresh working reagent was prepared prior to each use by combining Reagent A (NaCO3, NaHCO3, BCA and sodium tartrate in 0.1M sodium hydroxide) and reagent B (4% CuS04.5H2O) at a ratio of 50:1 (5 mL Reagent A to 100 µL Reagent B). The assay was run by mixing 50 µL of cell lysate (diluted in distilled water as required) with 1 mL of working reagent prior to a 30 minute incubation on a shaking heating block at 60o C to start the reaction. When complete the reaction was stopped by placing the samples on ice until cool and the absorbance read at 562 nM against a water blank. Protein concentration was determined using a standard curve by measuring the absorbance of known concentrations of BSA (0-250 µg/mL) under the same conditions.
2.2.4 SDS-PAGE
2.2.4.1 Cell lysate preparation.
U937 samples for western blot were prepared in the following manner. All medium within the wells were pippeted up and down to dislodge the cells into suspension before transfer into 1.7 mL centrifuge tubes. Cells were centrifuged for 15,000g, 0◦C for 5 minutes in order to pellet cells. The supernatant was discarded and cells re-suspended in sterilized ice cold PBS before being centrifuged at 15,000 g, at 0oC for 5 miniutes in order to wash the cells. The supernatant was discarded and 150 µL of ice-cold lysis buffer (containing protease inhibitor) was added to each centrifuge tube and left on ice for 25 miniutes in order to ensure lysation. Cell lysate was stored at -80oC degrees until analyzed.
2.2.4.2 SDS-polyacrylamide gel electrophoresis
The required amount of sample as determined by protein concentration (generally 50 µg at volumes ~30-50 µL) was aliquoted into 1.7 mL centrifuge tubes. 400 µL of ice-cold acetone was added to each tube and centrifuged for 15,000, 0oC for 10 minutes to pellet the cells. The supernatant was discarded and the tubes were left in a fume hood for any residual acetone to evaporate. The pellet was re-suspended in fresh cracker buffer containing 2% β-mercaptoethanol to a final protein concentration of 2µg protein per µL. The samples were then heated in a heating block at 90oC for 2 minutes to denature the proteins. The samples were centrifuged for 5 minutes at 20,000 g. 5 µL of pre-stained molecular weight marker mix (Fermentas International Inc, Ontario, Canada) and 25 µL/well of sample were loaded into the gel wells. The gel (4-12% Bis-Tris Gel, Invitrogen, California, USA) was run at 100V at 300mA in 1x MOPS buffer until the dye had run past the wells, and 200V at 300mA thereafter until the dye had reached the bottom of the gel.
2.2.5 Western blot analysis
2.2.5.1 Protein transfer
Proteins separated on the SDS-PAGE gel were transferred onto a nitrocellulose membrane using either wet or semi-dry transfer protocols. For the wet method transfer occurred over 800 minutes at 70V in a water cooled tank transfer electrophoresis unit (TE22, Hoefer, USA) containing the transfer buffer. For the semi-dry method transfer occurred at 1.3A, 25V for 10 minutes using the Trans-Blot® Turbo™ Transfer System (Bio-Rad Laboratories, California, USA) following the manufactures instructions.
The following steps were all undertaken on a rocking platform mixer (Ratex Instruments, Australia). Once transfer had been completed the nitrocellulose membrane was rinsed with nano-pure water and incubated with 0.01% Ponceau S stain for 1 minute to check transfer efficiency. Efficient transfer showed distinct pink columns forming at the location of each well. The membrane was then blocked using 5% TBSM for 1.5-2 hours with the TBSM being replaced every 30 minutes. During this time the Ponceau S stain completely disappeared.
2.2.5.2 CD36 immunostaining
Following blocking the nitrocellulose membrane was incubated with rabbit polyclonal affinity purified antibody raised against human CD36 (NB400-145, Novus Biologicals Inc., USA) for 1.5 hours diluted to 1:1,000-2000 in 1% TBSM. The nitrocellulose membrane was then washed 5 times for 5 minutes in TBS before undergoing incubation for 1 hour with the secondary antibody; goat polyclonal anti-rabbit IgG, conjugated to hydrogen peroxidase (HRP) (NB730-H, Novus Biologicals Inc., USA) diluted to 1:2,000. Incubation was followed by 5 lots of 5 minute washes in TBS before being briefly rinsed with nano-water prior to visualization.
2.2.5.3 Visualization
Visualization of the antibody staining was achieved via chemiluminescence signaling corresponding to the location of the HRP-conjugated secondary antibody. The signal was produced by incubating the nitrocellulose membrane with Supersignal West Dura Chemiluminescence (TerumoScientific, USA) substrates A & B, mixed at a ratio of 1:1. Images containing the signal were obtained at high resolution, without filter or light, using the time-lapse auto-exposure setting on a Syngene Chemigenius-2 bioimaging system (USA). The images were then analyzed for band intensity using GeneSnap (Syngene, USA) and ImageJ64 1.42q.
2.2.5.4 Nitrocellulose membrane stripping and β-Actin probing
Re-probing of the nitrocellulose membrane for β-actin was always subsequent to visualization of CD36 during which time the nitrocellulose membrane was kept in TBS at 4◦C. Prior to re-probing for β-actin the nitrocellulose membrane was stripped of all previous antibodies via incubation with stripping buffer (2% SDS, 50mM Tris, 100µM β–MEtOH, pH 6.8, heated to 50◦C on a water bath) for 20 minutes, followed by 5 lots of 5 minute washes in TBS. Reprobing for β-actin was done directly after stripping without blocking.
Once stripping was completed the nitrocellulose membrane was incubated with mouse monoclonal antibody raised against β-actin (A5316, Sigma-Aldrich Chemical Co., USA) for 1.5 hours diluted to 1:7,500 in 1% TBSM. The nitrocellulose membrane was then washed 5 times for 5 minutes in TBS before undergoing incubation for 1 hour with the secondary antibody; anti-mouse IgG Peroxidase-linked NA931 (GE Healthcare, UK) diluted to 1:7,500 in 2% TBSM. Incubation was followed by 5 lots of 10 minute washes in TBS before being briefly rinsed with nano-pure water prior to visualization.
2.2.6 Flow cytometry determination of U937 CD36 cell surface expression
Flow cytometry is a powerful technique that is able to analyze the fluorescence of individual cells passing through a laser light source. The fluidic systems within the flow cytometer uses hydrodynamic focusing to arrange the particles in a sample into a straight stream that passes through the light source. The flow cytometer then collects light scattering and fluorescent emission information about the particle to reveal information about its properties. The flow cytometer contains two separate detectors, the Forward Scatter Channel (FSC) which is offset 20 degrees from the beam axis, and the Side Scatter Channel which is offset 90 degrees. The patterns generated from these two detectors are unique for every particle and provides information of particle size, granular content, while also allowing identification of live cells and cellular debris.
Cells were extracted from experimental wells and centrifuged in a pre-cooled centrifuge tube (4oC) for 5 minutes at 2000 rpm. The supernatant was then discarded and the cells re-suspended in 245 µL PBS + 0.5% BSA. 5 µL of Purified Mouse Anti-Human CD36 primary antibody (BD Biosciences) was added and incubated on ice for 20 minutes (final concentration: 10µg/mL mouse anti-human CD36). The cells were then centrifuged for 5 minutes at 2000 rpm before being re-suspended in 190 µL PBS +0.5% BSA and 10 µL of secondary antibody Polyclonal Goat Anti-Mouse Immunoglobulins/RPE Goat F(ab’)2 added. Incubation on ice was allowed to occur for 20 minutes before the cells where centrifuged 5 minutes at 2000 rpm and brought back up in 1mL PBS +0.5% BSA. 10,000 events were then analysed on the FL2 filter using the BD Biosciences Accuri C6 Flow Cytometer. A primary antibody control (IgMk) was used also to ensure that all labeling detected was seen purely due to CD36 antibody binding.
2.2.7 Statistical analysis
The results presented in this thesis were graphed and statistically analyzed by GraphPad Prisim version 6.01 for Windows (GraphPad Software, San Diego, California, USA). Significance was confirmed via either, one-way or two-way analysis of variance (ANOVA). Significance levels are indicated as: (*) p≤ 0.05, (**) p≤ 0.01 and (***) p≤ 0.001. Unless stated otherwise results presented within this thesis are shown in triplicate from at least three representative experiments. The graphs are presented as the mean and the standard error of the mean (SEM) was calculated from triplicate samples in most cases.
3. RESULTS
3.1 Characterising the expression of CD36
3.1.1 The U937 cell line produces detectable basal levels of CD36
Prior research exploring the down regulation of CD36 has focused mostly on the down regulation of this scavenger receptor after an up regulation event (Draude & Lorenz, 2000; J. Han et al., 2000). In contrast this research instead focuses on the down regulation of CD36 from the basal levels that naturally occur in the U937 cell line. Before research could commence it first had to be determined at what levels basal CD36 could be detected using western blotting protocols. To determine this U937 cells were incubated in RPMI-1640 with 5% FBS for 24 hours. Due to basal CD36 levels being the only ones of interest, cells were incubated solely RPMI-1640 media with foetal calf serum. Cell lysate was then immunoblotted for CD36 with β-actin used as a loading control. It was found that a strong CD36 band could be detected at the ~100 kDa mark, with a weaker band at ~88 kDa (Figure 3.1.1). Different bands represent the different glycosylation isoforms and maturity levels of the CD36 protein. The 88 kDa band is reported to be incompletely glycosylated CD36 located in the Golgi apparatus, whereas the fully glycosylated 100 kDA band is mature CD36 residing on the plasma membrane (Alessio et al., 1996). The strength of the bands observed was considered to be suitable for detecting changes in basal CD36 levels upon treatment. It was decided that the main focus of this research would be on the 100 kDa isoform due to the strength of the band, and that cell surface CD36 is likely responsible for oxLDL binding leading to foam cell formation.
100 kDa
88 kDa
Figure 3.1.1 The U937 cell line produces detectable basal levels of CD36
U937 cells (0.5×106 cells/mL) were incubated at 37oC for 24 hours in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. Quantitative densitometry of western blots shows a strong band at ~100 kDa and a weaker band at ~88 kDa.
3.1.2 Triton X-100 as a lysing agent
Before further research could take place, appropriate experiments had to be under taken in order to determine an appropriate lysing agent for efficient use with the U937 cell line. Previous research using human monocyte derived macrophages (HMDM) in the Free Radical Biochemistry (FRB) lab group used a HEPES and NaCl based lysing agent. Initial experiments showed that this failed to efficiently lyse U937 cells. To overcome this Triton X-100 was considered for use. Triton X-100 is a nonionic surfactant detergent successfully used previously in working with the U937 cell line. In order to re-examine its use here U937 cells were incubated with increasing concentrations of Triton X-100 of between 0 and 1.0% concentration for 10 minutes at 37oC. Cell viability and lysation was determined via a light microscope in order to determine the appropriate amount of Triton X-100 required for complete lysation. Cell images of the Triton X-100 treatments showed that 0.05% Triton X-100 was the minimum concentration required to lyse 100% of viable cells (Figure 3.1.2).



Control
0.01% Triton X-100
0.1% Triton X-100


0.5% Triton X-100
1.0% Triton X-100
Figure 3.1.2 0.5% TritonX-100 effectively lysis cells
Images of U937 cells (0.5×106 cells/mL) after incubation with 0–1% Triton X-100 for 10 minutes at 37oC. Cells were viewed in tissue culture plates through an inverted microscope (20x magnification). The images were taken using a LEICA DMIL microscope with a LEICIA DFC290 camera. The population density is a 500,000 cells per mL.
3.1.3 Effect of PMA of U937 expression
After confirmation that the U937 cell line produced detectable basal levels of CD36 it was of interest to further characterise CD36 expression. One aim was to see whether expression could be increased and if so to what extent. CD36 expression is known to increase as monocytes differentiate into macrophages (Chanput, Peters, & Wichers, 2015). Phorbol-12-myristate-13-acetate (PMA) is commonly used as a stimuli in the differentiation process and was considered an appropriate choice to use here. In order to examine the effect of this differentiation agent on CD36 levels U937 cells were incubated in RPMI-1640 with 5% FBS and 25nM PMA over a 24 hour time course. After incubation with PMA cell lysate was immunoblotted for CD36 with β-actin used as a loading control.
Results showed that at the 6 hour time point there appeared to be an increase in CD36 expression to 129.66% of control (Figure 3.1.3). This increase however was non-significant and had dropped down to 87.93% of control by the 12 hour time point. CD36 expression then held relatively steady till time course completion at 24 hours at which it was found to be present at 92.62% of control. Overall the results showed that at 25nM PMA caused no significant change in CD36 expression over a 24 hour time course.
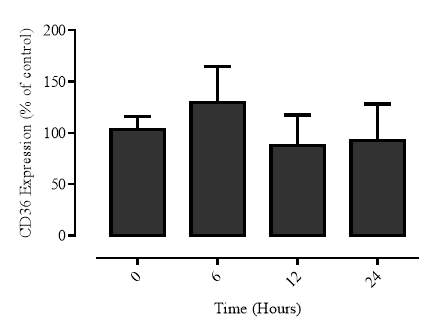
A
B
100 kDa
β-actin
Time (Hours) 0 6 12 24


Figure 3.1.3 PMA has a limited effect on U937 expression
U937 cells (0.5×106 cells/mL) were treated with 25nM of PMA and incubated at 37oC over a 24 hour time course in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of duplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (**) p ≤ 0.001. B) Quantitative densitometry of western blots shows PMA having a limited effect on CD36 relative to controls.
3.2 Characterization of CD36 down regulation via 7,8-NP
3.2.1 7,8-NP reduces the expression of the CD36 scavenger receptor in the U937 cell Line
Previous research has shown that the macrophage derived antioxidant 7,8-dihydroneopterin (7,8-NP) caused a decreased in CD36 expression at concentrations up to 250µM in HMDM cells (S. P. Gieseg, Z. Amit, Y.-T. Yang, A. Shchepetkina, & H. Katouah, 2010). While the U937 cell line is considered to be a good model for researching monocytes, 7,8-NP induced down regulation needed to be confirmed here. To achieve this U937 cells were incubated for 24 hours with 7,8-NP concentrations ranging from 0-150µM. Cell lysate was then immunoblotted for the CD36 protein with β-actin used as a loading control. Down regulation was observed beginning at 50µM 78-NP with CD36 expression found to be 54.77% of control (Figure 3.2.1). Expression then decreased further at both 100µM and 150µM concentrations (44.43% and 42.73% of control respectively). Significance was observed at all three data points, with the highest level of significance occurring at 150µM. No significant changes were observed between the different concentrations. The strength of this down regulation showed that the effect of 7,8-NP on CD36 in the U937 cell line was stronger to that seen with HMDM’s and that 150µM 7,8-NP was the most appropriate concentration to use in further experimentation due to it having the most significant effect.
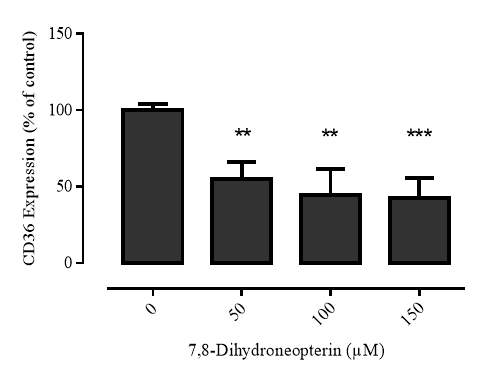
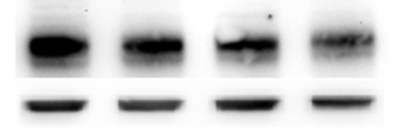
A
B
100 kDa
β-actin
7,8-NP (µM) 0 50 100 150

Figure 3.2.1 7,8-NP reduces the expression of the CD36 scavenger receptor in the U937 cell line
U937 cells (0.5×106 cells/mL) were treated with 0-150µM 7,8-NP and incubated at 37oC over a 24 hour time course in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of triplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots shows 7,8-NP reduces CD36 expression between 50-150µM relative to controls.
3.2.2 7,8-NP induced down regulation occurs in both adherent and suspension well plates
Two common cell culture plates used in experimental work are adherent and suspension plates. In adherent plates cells will interact closely with the plate, forming an adherent layer on the well surface, where as in suspension plates cells are suspended within the media minimising well interaction. It is possible that the type of cell culture plate used may have an effect on the morphology and differentiation of U937 cells, and subsequently production of the heavily glycosylated 100 kDa CD36 band. This is more likely to occur when adherent plates are used due to closer interaction between the cells and the plate, which may be recognised as a foreign body and trigger the differentiation process. In order to rule out any effect due to the type of plate used on CD36 levels, and the subsequent effects of 7,8-NP, both adherent and suspension plates were examined for use in CD36 down regulation experiments. U937 cells were incubated with increasing concentrations of 7,8-NP ranging from 0-150µM for 24 hours in both adherent and suspension plates. Subsequently the cells were washed and lysed. Cell lysate was immunoblotted for CD36 with β-actin used as a loading control.
It was found that CD36 could be detected at strong basal levels when using both types of plates and that a concentration related decrease could be observed with increasing concentrations of 7,8-NP in both cases (Figure 3.2.2). These results show that any U937 interaction with the type of plate use has no or a very limited effect on basal CD36 levels and 7,8-NP induced down regulation. As such all further experimentation looking at this effect will take place using suspension well plates owing to lesser cell-plate interaction and easier use in extracting cells from the wells.

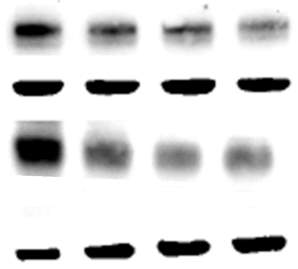
100 kDa
β-actin
Adherent
100 kDa
β-actin
Suspension
Figure 3.2.2 7,8-NP down regulation can be observed using both adherent and suspension plates
U937 cells (0.5×106 cells/mL) were treated with increasing concentrations of 7,8-NP (0-150µM) and incubated at 37oC for 24 hours in RPMI-1640 with phenol red using both adherent and suspension well plates. Cell lysate was assessed for CD36 protein expression via western blot. Figure shows the quantitative densitometry of western blots and that increasing 7,8-NP concentration reduces CD36 expression relative to controls in both plate types.
3.2.3 7,8-NP down regulation can be observed using both wet and semi-dry protein transfer protocols
Western blotting involves the transfer of proteins after polyacrylamide gel electrophoresis over to a membrane suitable for antibody probing such as nitrocellulose. Two commonly used transfer techniques (wet and semi-dry) were explored for their suitability in experimental work looking at the 7,8-NP induced down regulation of CD36. The wet transfer technique system uses a larger volume of transfer buffer and is run over a longer time period, whereas the semi-dry transfer system uses less buffer and is run over a shorter time. The wet transfer system is usually considered to be better suited for larger molecular weight proteins, whilst the semi-dry method is thought to be more ideal for smaller proteins. To explore any differences in their suitability for this work U937 cells were incubated with increasing concentrations of 7,8-NP ranging from 0-150µM with protein transfer occurring using either a wet or semi-dry protocol. It was found that both systems were able to detect 7,8-NP induced down regulation of CD36 to a similar extent as confirmed via antibody probing (Figure 3.2.3). Both systems also transferred similar amounts of protein from the SDS page gel onto the nitrocellulose membrane as detected via both ponceau staining of the nitrocellulose membranes after transfer, along with coumassise blue staining of the SDS gel itself (results not shown). However the semi-dry method was found to produce a more reliable transfer with almost no air bubbles forming during the transfer process compared to the wet transfer method. Overheating of the nitrocellulose membranes was also cut back when the semi-dry method was used. Due to these factors the semi-dry system was the method of choice for all future transfers.

100 kDa
β-actin
Wet
100 kDa
β-actin
Semi-Dry
Figure 3.3.3 7,8-NP down regulation can be observed using both wet and semi-dry transfer protocols
U937 cells (0.5×106 cells/mL) were treated with increasing concentrations of 7,8-NP (0-150µM) and incubated at 37oC for 24 hours in RPMI-1640 with phenol red using both wet and semi-dry transfer methods. Cell lysate was assessed for CD36 protein expression via western blot. Figure shows quantitative densitometry of western blots and that increasing 7,8-NP concentration reduces CD36 expression relative to controls in protein transfer systems.
3.2.4 Time course of 7,8-NP induced CD36 down regulation
Once CD36 down regulation was confirmed in the U937 cell line a time-course was performed in order to investigate the speed at which 7,8-NP has its effect. To achieve this U937 cells were incubated with 150µM 7,8-NP over a 24 hour time course. During incubation the cells were extracted from the wells at 0, 6, 12 and 24 hours and lysed in the presence of protease inhibitor. Cell lysate was kept at -80oC until analysis. Once the time course was complete the cell lysate was then immunoblotted for the CD36 protein with β-actin used as a loading control.
Down regulation was observed beginning at 6 hours with CD36 expression found to be at 55.12% of control (Figure 3.2.2). CD36 expression then continued to decrease at both 12 and 24 hour time points (43.56% and 29.16% of control respectively). Change compared to control was significant at all time points, being the most significant at the 24 hour time point. No significant changes were observed between the different time points. The results suggest that CD36 down regulation occurs rapidly within the first six hours of incubation with 7,8-NP but continues on to some extent to the 24 hour time point.
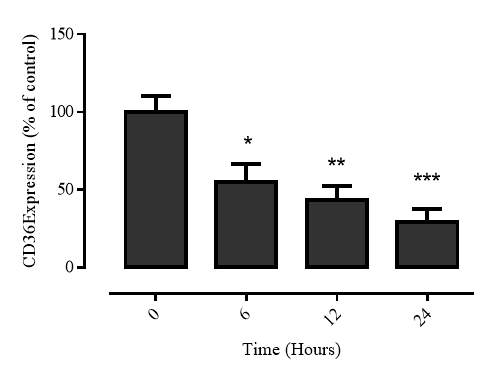

A
B
100 kDa
β-actin
Time (Hours) 0 6 12 24

Figure 3.2.4 7,8-NP down regulates CD36 over 24 hours
U937 cells (0.5×106 cells/mL) were treated with increasing concentrations of 7,8-NP (0-150µM) and incubated at 37oC for 24 hours in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of triplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots shows 150µM 7,8-NP decreasing CD36 expression over 24 hours relative to controls.
3.2.5 Recovery of CD36 expression after 7,8-NP removal
Once the 7,8-NP induced down regulation of CD36 had been established the next step was to examine the recovery of CD36 after 7,8-NP removal. Investigation into the recovery of the cells after treatment is an important factor into the characterization of 7,8-NP’s effect on CD36. It also provides evidence as to whether or not cells are suitable for further experimentation after initial down regulation. Previous work examining the recovery of HMDMs after the removal of 7,8-NP was limited and only examined recovery up to a six hour time point (Schepetkina, 2013). Here we look at recovery up to 48 hours after 7,8-NP removal. U937 cells were incubated for 24 hours with 150µM 7,8-NP in a 75 cm3 cell culture flask in order to allow CD36 down regulation to take place. The cells were then centrifuged at 500x g to pellet the cells and the supernatant discarded to remove 7,8-NP. The cells were then re-suspended in fresh media before transfer into a 24 well suspension plate and incubated at 37oC. Cells were extracted from the wells at 0, 12, 24 and 48 hour time points and lysed in the presence of protease inhibitor and kept at -80oC until analysis. Once the time course was complete the cell lysate was then immunoblotted for the CD36 protein with β-actin used as a loading control.
The results show that after a 24 hour incubation period with 150µM 7,8-NP CD36 levels were down-regulated to a significant 50.22% of control (Figure 3.2.5). Recovery was then observed 12 hours after 7,8-NP removal where CD36 expression had recovered to a non-significant 63.17% of control, and 24 hours after removal CD36 expression had recovered to a significant 96.02% of control. Between the 24 and 48 hour time points CD36 expression began to decrease again to 30.91% of control at 48 hours. Recovery of CD36 between the 12 and 24 hour time points, while showing that recovery does occur, limits time for any subsequent experimental work to between 0 and 12 hours after removal to ensure levels of CD36 are still down regulated.

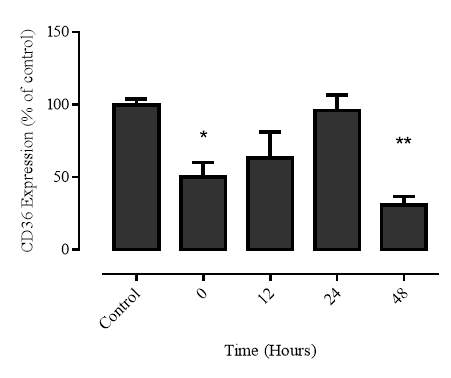
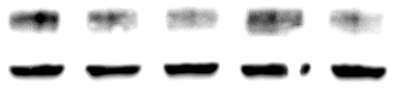
A
B
100 kDa
β-actin
Time (Hours) Control 0 12 24 48
Figure 3.2.5 CD36 expression recovers 24 hours after 7,8-NP removal
U937 cells (0.5×106 cells/mL) were treated 150µM 7,8-NP and incubated at 37oC over 24 hours in RPMI-1640 with phenol red to allow for down regulation to occur. 7,8-NP was then removed and the cells plated for an additional 48 hour time course. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of triplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots shows 7,8-NP CD36 expression recovering 24 hours after 7,8-NP removal relative to controls.
3.2.6 Cellular viability of the recovery experiment
During the recovery experiment the U937 cells underwent down regulation via 7,8-NP for 24 hours before being plated for up to an additional 48 hours. The viability of this experiment was investigated at each time point to ensure that the results observed were not influenced by the loss of cellular viability over the experimental time course. At each time point (0, 12, 24 and 48 hours) prior to full cellular extraction and lysation, 50 µL of cells were taken from each well and incubated with 0.4% trypan blue at a ratio of 1:1 for 2 minutes before viability analysis using a heamocytometer. Results showed that the recovery experiment had very little effect on cell viability (Figure 3.2.6). Viability immediately after 7,8-NP removal was found to be at 96.37%. This level then held relatively steady over the entire time course with viability after 48 hours measured at 95.11%. No significant changes were observed between the different time points.
Cells were also viewed under a light microscope at 20x magnification and images taken to observe morphological changes. Changes in cell morphology during 7,8-NP induced down-regulation and after removal appear to be limited. Control cells show a typical spherical morphology which continued on through to 48 hours after 7,8-NP removal.
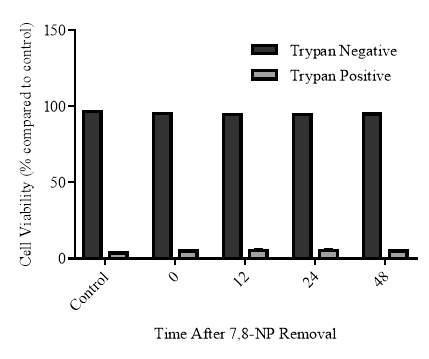
A
B



Control
0 Hours
12 Hours


24 Hours
48 Hours
Figure 3.2.6 CD36 expression recovers 24 hours after 7,8-NP removal
U937 cells (0.5×106 cells/mL) were treated with 150µM of 7,8-NP at 37oC over 24 hours. 7,8-NP was then removed and cells incubated for a further 48 hour time course. A) Cell viability was measured by tyrpan blue and the data was expressed as a percentage of the respective control (cell only control). Results are displayed as mean ± SEM of triplicates from a single experiment, representative of three separate experiments. Significance levels are indicated as: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B. Images of U937 cells during each time point of recovery. B) Cells were viewed in tissue culture plates through an inverted microscope (20x magnification). The images were taken using a LEICA DMIL microscope with a LEICIA DFC290 camera. The population density is a 500,000 cells per mL.
3.2.7 Effect of neopterin on CD36 expression in the U937 cell line
Neopterin is the oxidised product of 7,8-NP, and like 7,8-NP is also a member of the pteridine family. Biologically it can be used to provide information on cellular immune activation and as an inflammatory disease marker (Murr, Widner, Wirleitner, & Fuchs, 2002). Once the down regulation effect of 7,8-NP was characterised in the U937 cell line it was of interest to measure any effect that neopterin may have on CD36 levels. To achieve this U937 cells were incubated with increasing levels of neopterin ranging from 0-100µM for 24 hours. Afterwards the cells were extracted from the wells and lysed. The cell lysate was then immunoblotted for the CD36 protein with β-actin used as a loading control.
The results show that in the 0-100µM concentration range neopterin had no significant effect of CD36 expression compared to control levels (Figure 3.2.7). At 25µM & 75µM neopterin CD36 expression was 8.14% and 19.43% lower than control respectively, while at 50µM and 100µM CD36 expression was 6.50% and 12.29% higher than control. The fact that none of these changes were found to be significant or followed a discernible pattern indicates that unlike 7,8-NP, neopterin has no effect on CD36 expression.
Cells were also viewed under a light microscope at 20x magnification and images taken to observe any morphological changes upon treatment. Changes in cell morphology during treatment with neopterin are limited with control cells showing a typical spherical morphology comparable to that seen up to and including 150µM neopterin (Figure 3.2.7).
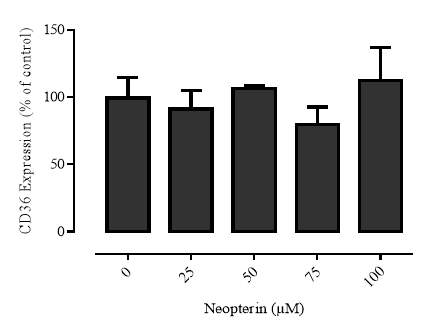
A

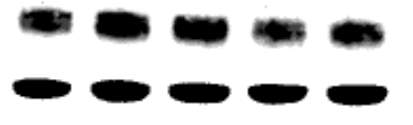
B
100 kDa
β-actin
Neopterin (µM) 0 25 50 75 100
Figure 3.2.7 Neopterin has a limited effect on CD36 expression
U937 cells (0.5×106 cells/mL) were treated with increasing concentrations of Neopterin (0-100µM) at 37oC over 24 hours in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of triplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots shows limited effect on CD36 expression decreasing with increasing neopterin concentration relative to controls.



Control
25µM Neopterin
50µM Neopterin


75µM Neopterin
100µM Neopterin
Figure 17 Images of U937 cells after treatment with increasing concentrations of neopterin
Images of U937 cells after treatment with increasing concentrations of neopterin for 24 hours. Cells were viewed in tissue culture plates through an inverted microscope (20x magnification). The images were taken using a LEICA DMIL microscope with a LEICIA DFC290 camera. The population density is a 500,000 cells per mL.
3.3 Transcriptional control of CD36
3.3.1 Effect of PPARγ inhibitor GW9662 on CD36 expression
Previous publications have shown that CD36 is under control of a heterodimer formed between the nuclear receptors PPARγ and RXR (S. Han & Sidell, 2002). A kinase binding site located on the PPARγ component makes it a likely target for exploration into the 7,8-NP induced down regulation pathway. Yet before this could commence it first had to be confirmed that CD36 was indeed under control of this heterodimer in the U937 cell line. To achieve this GW9662, an irreversible antagonist of PPARγ was chosen for use. GW9662 selectively and covalently modifies the CYS285 residue of PPARγ, and is 10- and 600-fold less potent in binding experiments using PPARα and PPARδ isoforms (Leesnitzer et al., 2002).
U937 cells were incubated for 24 hours with increasing concentrations of GW9662 ranging from 0-15µM, with cells only used as a negative control and 150µM 7,8-NP as a positive control. At the 24 hour time point the cells were extracted and lysed. The cell lysate was then immunoblotted for the CD36 protein with β-actin used as a loading control. GW9662 significantly decreased CD36 expression beginning at 5µM (66.47% of control) with a similar level of effect seen at 10µM (72.69% of control) (Figure 3.3.1). 15µM GW9662 decreased CD36 expression further still (41.57% of control). Change compared to control was significant at all concentrations, being the most significant at 15µM. No significant changes were observed between the different concentrations. 150µM 7,8-NP alone decreased CD36 expression to 60.89% of control, comparable most to that seen with 5µM. The results confirm that CD36 is under control of PPARγ, and also our choice of PPARγ as a target in the exploration of the 7,8-NP induced down regulation pathway.

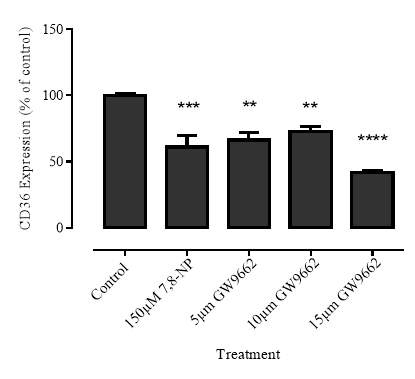


A
B
100 kDa
β-actin
Figure 3.3.1 PPARγ inhibitor GW9662 decreases CD36 expression
U937 cells (0.5×106 cells/mL) were treated with increasing concentrations of GW9662 (0-15µM) at 37oC over 24 hours in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of triplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots shows CD36 expression decreasing with increasing GW9662 concentration relative to controls.
3.3.2 Effect of GW9662 on cellular viability
The PPARγ transcription factor alongside its role in CD36 regulation, is involved in controlling a wide variety of genes over several pathways related to lipid metabolism. These include fatty acid transport, catabolism and storage (Hardwick, Osei-Hyiaman, Wiland, Abdelmegeed, & Song, 2010). The use of the GW9662 inhibitor would interfere with these processes with a possible negative effect on cellular viability. To confirm the reliability of the results of the previous experiments cell viability was measured using the propidium iodide assay. This assay can determine the ratio of alive cells which have excluded the dye (PI negative) to non-viable cells (PI positive). Cells were incubated for 24 hours with increasing concentrations of GW9662 of between 5-15µM in RPMI-1640 without phenol red as the use of this colored pH indicator has the potential to interfere with assay results. Results show that all treatments had a limited effect on cellular viability with all GW9662 concentrations used non-significantly reducing viability in a range of between 1.56-5.75% (Figure 3.3.2). The low impact that GW9662 had on viability, even at the higher concentrations, indicate cell viability loss was most likely not a factor in the GW9662 triggered decrease in CD36 expression.
Changes in cell morphology after GW992 treatment was examined using a light microscope (Leitz Wetzlab, Germany). Changes in cell morphology during treatment with GW9662 are limited with control cells showing a typical spherical morphology comparable with that seen up to and including 15µM GW9662 (Figure 3.2.2).
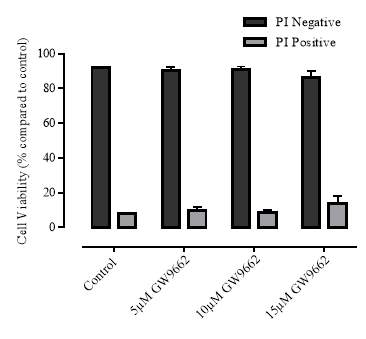
A
B


Control
5µM GW9662


10µM GW9662
15µM GW9662
Figure 3.3.2 Effect of GW9662 on cellular viability using propidium iodide
U937 cells (0.5×106) were treated with between 0-15µM GW9662 at 37oC over 24 hours in non-phenol red RPMI-1640. A) Cell viability was measured by PI and the data was expressed as a percentage of the respective control (cell only control). Results are displayed as mean ± SEM of triplicates from a single experiment, representative of three separate experiments. Significance levels are indicated as: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Images of U937 cells after treatment with increasing concentrations of GW9662 for 24 hours. Cells were viewed in tissue culture plates through an inverted microscope (20x magnification). The images were taken using a LEICA DMIL microscope with a LEICIA DFC290 camera. The population density is a 500,000 cells per mL.
3.3.3 Effect of GW9662 in conjunction with 7,8-NP on CD36 expression
Once it was confirmed that both 7,8-NP and GW9662 were able to down regulate observed levels of CD36, both were given in conjunction to U937 cells to determine if any additive effect on CD36 could be seen. To achieve this U937 cells were incubated for 24 hours with 5µM GW9662 in combination with 150µM 7,8-NP. A 5µM concentration of GW9662 was chosen as this level was shown to down-regulate CD36 levels to a similar extent as 150µM 7,8-NP. GW9662 and 7,8-NP were also incubated with the cells in isolation to act as positive controls. At the 24 hour time point the cells were extracted from the wells and lysed. The cell lysate was then immunoblotted for the CD36 protein with β-actin used as a loading control. The results show that while both treatments down regulated observed CD36 levels to comparable levels, no additive effect could be seen. 7,8-NP in conjunction with GW9662 down regulated CD36 expression to 62.00% of control. This result is comparable to 7,8-NP (58.55%) and GW9662 alone (61.49%) (Figure 3.3.3). This indicates that if 7,8-NP is having its effect via PPARγ that a maximum level of down regulation can be reached where any additional treatment will have an un-proportional effect.
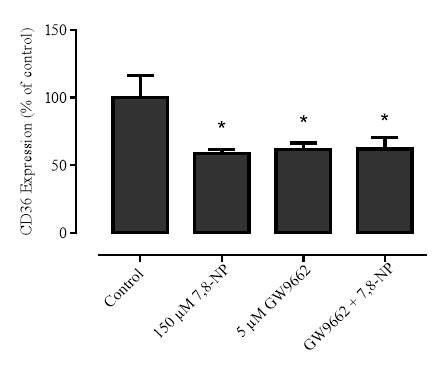
A
B


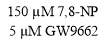

100 kDa
β-actin
Figure 3.3.3 GW9662 and 7,8-NP do not have an additive effect on 7,8-NP expression
U937 cells (0.5×106 cells/mL) were treated with 5µM GW9662 in conjunction with 150µM 7,8-NP at 37oC over 24 hours in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of quadruplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots shows no additive effect on CD36 expression with both GW9662 and 7,8-NP relative to controls.
3.3.4 Effect of increasing concentrations of cycloheximide on cellular viability
Cycloheximide produced by the bacterium Streptomyces griseus acts as an inhibitor of protein synthesis in Eukaryotic organisms by interfering with the translocation process (Jones & Cushman, 1989). It is used in determining protein half-life via the use of time-course experiments in conjunction with western-blotting. This enables the detection of changes in protein bound signal over time. Due to its effect on protein synthesis the use of cycloheximide is likely to have an effect on cellular viability at higher levels that would confound any experimental results. In order to examine what levels are suitable for experimental work involving the U937 cell line a cell viability time course was examined using various concentrations of cycloheximide. U937 cells were incubated for 24 hours with increasing concentrations of cycloheximide ranging between 0-10µM in RPMI-1640 without phenol red. At the 24 hour time point cells were extracted from cells and incubated with PI in the dark for 10 minutes before viability was measured. Results showed that increasing concentrations of cycloheximide had a negative effect on cellular viability (Figure 3.3.4). 2-5µM cycloheximide decreased viability in a range of 6.15-7.87% lower than control after 24 hours, however this decrease was non-significant. 10µM cycloheximide had a bigger impact, decreasing cellular viability to a significant 22.99% lower than that of control. No significant changes were observed between the different concentrations
Changes in cell morphology after Cycloheximide treatment were examined using a light microscope (Leitz Wetzlab, Germany). U937 cells in the absence of cycloheximide showed a typical spherical morphology. Cells treated with 2µM cycloheximide appear lower in density but still display a healthy morphology albeit with a small amount of debris present. At 5µM continuing on to 10µM density decreased and cellular debris increased with pronounced blebbing becoming apparent. (Figure 3.3.4)
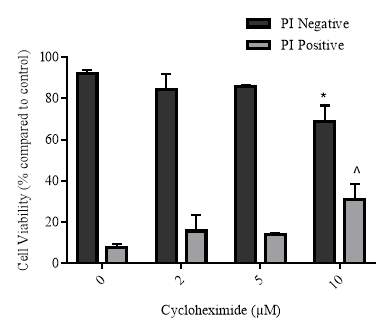
A
B

2µM Cycloheximide
Control
5µM Cycloheximide
10µM Cycloheximide
Figure 3.3.4 Effect of increasing concentrations of cycloheximide on cellular viability
U937 cells (0.5×106 cells/mL) were treated with 0-10µM of cycloheximide at 37oC over 24 hours in non-phenol red RPMI-1640. A) Cell viability was measured by PI and the data was expressed as a percentage of the respective control (cell only control). Results are displayed as mean ± SEM of triplicates from a single experiment, representative of three separate experiments. Significance levels are indicated as: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Images of U937 cells after treatment with increasing concentrations of cycloheximide for 24 hours. Cells were viewed in tissue culture plates through an inverted microscope (20x magnification). The images were taken using a LEICA DMIL microscope with a LEICIA DFC290 camera. The population density is a 500,000 cells per mL.
3.3.5 Effect of cycloheximide on CD36 expression
After completion of the cell viability experiment a concentration of 1µM cycloheximide was decided as best suitable for use. Next a cycloheximide time course was undertaken in order to measure the natural half-life of CD36 and to explore whether 7,8-NP additionally has any effect on promoting CD36 degradation.
U937 cells were plated in 24 well suspension plates and incubated with either 1) 150µM of 7,8-NP, or 2) 150µM of 7,8-NP in conjunction with 1µM cycloheximide. After incubation cells were extracted from the wells at 0, 3, 6 and 9 hour time points, lysed in the presence of protease inhibitor and kept at -80oC until analysis. Once the time course was complete the cell lysate was then immunoblotted for the CD36 protein with β-actin used as a loading control. A 3 hour incubation with 1µM cycloheximide triggered a decrease in CD36 expression to 83.61% of control levels while cycloheximide + 7,8-NP triggered a decrease to 73.61% of control levels. At 6 hours CD36 expression had further decreased with both cycloheximide, and cycloheximide + 7,8-NP (49.93% and 53.53% respectively). While at 9 hours the detected CD36 levels were 32.91% and 36.19% respectively. Significance was detected for both cycloheximide and cycloheximide + 7,8-NP treatments after 6 and 9 hours of incubation but not at 3 hours. There was no significance between treatments at each respective time point. The results indicate that the half-life for CD36 is at around 6 hours, and that as there was lack of any significant difference between the treatments, that 7,8-NP is not enhancing the degradation of CD36 but rather is effecting CD36 transcription, complimenting previous mRNA data (Shchepetkina, 2013).
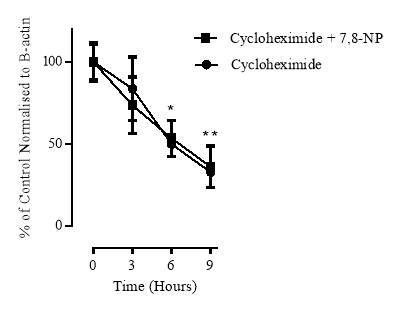
A

B
100 kDa
β-actin
Cycloheximide

Time: 0 3 6 9
C
Cycloheximide + 7,8-NP
100 kDa
β-actin

Time: 0 3 6 9
Figure 3.3.5 Effect of cycloheximide on CD36 expression
U937 cells (0.5×106) were treated with 1µM of cycloheximide at 37oC over 12 hours in non-phenol red RPMI-1640. A) Results were normalised by β-actin and displayed as mean ± SEM of quadruplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control B) Quantitative densitometry of western blots shows effect of 1µM of cycloheximide on the degradation of CD36 over 9 hours. C) Quantitative densitometry of western blots shows effect of cycloheximide + 7,8-NP on the degradation of CD36 over 9 hours. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001.
3.3.6 Cellular viability of the cycloheximide experiment
During the 1µM cycloheximide time course the viability of this experiment was investigated at each time point to ensure that the results observed were not influenced by the loss of cellular viability. At each time point (0, 3, 6 and 9 hours) prior to full cellular extraction and lysation 50 µL of cells were taken from each well and incubated with 0.4% trypan blue at a ratio of 1:1 for 2 minutes before analysis using a heamocytometer. Results show both cycloheximide, and cycloheximide + 7,8-NP treatments had a limited effect on cellular viability over the 9 hour time course (Figure 3.3.6). 1µM cycloheximide decreased viability to a non-significant 6.86% lower than control by the 9 hour time point where as cycloheximide + 7,8-NP decreased viability to a similar 5.36 % of control. The results indicate that loss of viability was not an issue in the cycloheximide experiment.
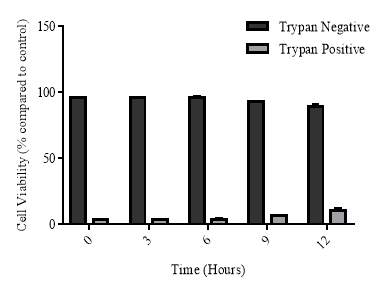
A
Cycloheximide
B
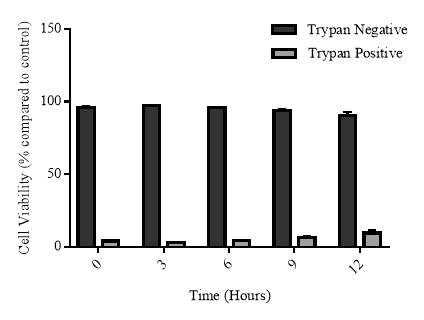
Cycloheximide + 7,8-NP
Figure 3.3.6 Effect of cycloheximide and cycloheximide+7,8-NP on U937 cell viability using trypan blue
U937 cells (0.5×106) were treated with 1µM of cycloheximide (A) or cycloheximide + 7,8-NP (B) at 37oC over 12 hours in non-phenol red RPMI-1640. Cell viability was measured by PI and the data was expressed as a percentage of the respective control (cell only control). Results are displayed as mean ± SEM of triplicates from a single experiment, representative of three separate experiments. Significance levels are indicated as: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001.
3.4 Effect of MAP kinase inhibitors on CD36 down regulation
3.4.1 Effect of JNK inhibition via SP600125 on 7,8-NP induced down regulation in the U937 cell line
Phosphorylation of PPARγ leads to a decrease in its CD36 transcriptional activity which in turn lowers CD36 protein expression (Adams et al., 1997; Hu et al., 1996). This phosphorylation effect makes protein kinase pathways a possible target for 7,8-NP induced down regulation. PPARγ’s kinase site has been shown to recognise and bind two different MAP kinases, c-Jun N-terminal kinase (JNK) and extracellular signal–regulated kinases (ERK) 1/2. To explore the effect on inhibiting the JNK MAP kinase pathway on CD36 expression the cell permeable and selective inhibitor SP600125 was used. SP600125 works by competing with JNK’s ATP binding site which halts the flow of the kinase cascade, thus preventing the signal from progressing (Bennett et al., 2001). U937 cells where pre-incubated with 50µM SP600125 in a 24 well suspension plate. 150µM 7,8-NP in conjunction with SP600125 was additionally added to measure whether any additive effect could be observed. 150µM 7,8-NP alone was used as a positive control and cells only as a negative control. At the 24 hour time point cells were extracted and lysed in the presence of a protease inhibitor. Cell lysate was immunoblotted for CD36 with β-actin used as a loading control.
Results showed that SP600125 did not block 7,8-NP’s effect on CD36 expression but rather was able to decrease CD36 expression to 32.65% of control alone (Figure 3.4.1). This decrease was more pronounced than the 7,8-NP control which down regulated CD36 expression to 64.86% of control. When given in conjunction SP600125 and 7,8-NP lowered CD36 expression to 42.61% of control. All results were significant.

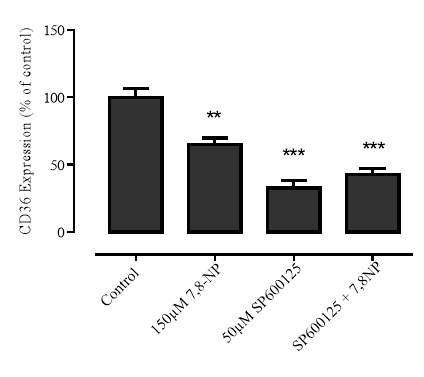
A
B
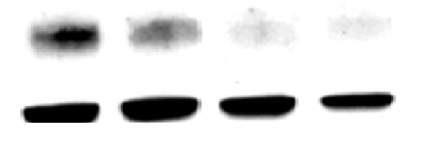
100 kDa
β-actin

150µM 7,8-NP
50µM SP600125
Figure 3.4.1 JNK inhibition via SP600125 does not alleviate CD36 down regulation
U937 cells (0.5×106 cells/mL) were treated with 150µM 7,8-NP, 50µM of SP600125, and both in conjunction at 37oC over 24 hours in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of triplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots showed that SP600125 did not rescue the effect of 7,8-NP on CD36 expression relative to controls.
3.4.2 Effect of JNK inhibition on cellular viability
JNK kinase cascades are involved in mediating the cellular apoptotic response to pro-inflammatory cytokines, genotoxins and environmental stress, along with having a role in the regulation of cell proliferation, survival and differentiation (C. Davies & Tournier, 2012). Use of the SP600125 inhibitor would interfere with these processes with possible negative effects on cellular viability. To confirm the reliability of the results of the previous experiment cell viability was measured using the propidium iodide assay. Cells were incubated for 24 hours with 50µM SP600125, along with 50µM SP600125 in conjunction with 150µM 7,8-NP, 150µM 7,8-NP alone and a cell only control. At the 24 hour time point cells were extracted and cell viability measured by PI using flow cytometry.
Results show that at 50µM SP600125 had a limited effect on U937 cellular viability. After a 24 hour incubation viability decreased to 7.16% lower than that of control (Figure 3.4.2). Even though this decrease was found to be significant, the low level of change means that it was unlikely to have had a major impact on the observed results. In contrast after incubation with 150µM 7,8-NP viability was 1.37% lower than control and 7,8-NP + SP600125 in combination 3.41% lower than control.
Changes in cell morphology after SP600125 treatment was examined using a light microscope (Leitz Wetzlab, Germany). Changes in cell morphology during treatment with SP600125 are non-existent with all cellular treatments showing a typical healthy morphology (Figure 3.4.2B). In treatments with SP600125 sharp yellow strands can be seen which appear to be the inhibitor precipitating out of solution.
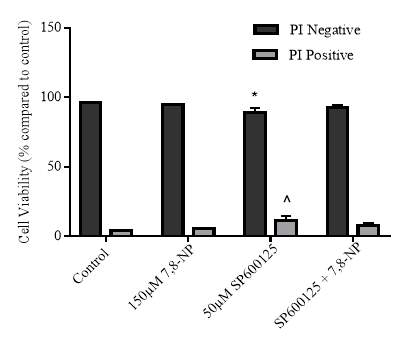
B
A
B


150µM 7,8-NP
Control


50µM SP600125
SP600125 + 7,8-NP
Figure 3.4.2 Effect of SP600125 on U937 cell viability using propidium iodide
U937 cells (0.5×106) were treated with 150µM 7,8-NP, 50µM of SP600125 and both in conjunction at 37oC over 24 hours in non-phenol red RPMI-1640. A.) Cell viability was measured by PI and the data was expressed as a percentage of the respective control (cell only control). Results are displayed as mean ± SEM of triplicates from a single experiment, representative of three separate experiments. Significance levels are indicated as: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Images of U937 cells after treatment with 150µM 7,8-NP, 50µM of SP600125 and both in conjunction for 24 hours. Cells were viewed in tissue culture plates through an inverted microscope (20x magnification). The images were taken using a LEICA DMIL microscope with a LEICIA DFC290 camera. The population density is a 500,000 cells per mL.
3.4.3 Effect of MEK inhibition via PD98059 on 7,8-NP induced down regulation in the U937 cell line
PPARγ’s kinase site, alongside recognising JNK, has also been shown to recognise and bind the ERK1/2 MAP kinase (Burns, 2007). To explore the effect on inhibiting the ERK1/2 MAP kinase pathway on CD36 expression the inhibitor PD98059 was used. PD98059 has its effect by selectively inhibiting the MAP kinase kinases MEK1 and MEK2 by binding to their inactive form (S. P. Davies, Reddy, Caivano, & Cohen, 2000). This prevents any activators from binding and effectively blocks downstream activation of ERK1/2 and the signalling cascade. U937 cells where pre-incubated with 25µM PD98059 before 150µM 7,8-NP was added to selected wells to measure whether any additive effect between the two could be observed. 150µM 7,8-NP in isolation was used as a positive control and cells only as a negative control. At the 24 hour time point cells were extracted, washed in PBS and lysed in the presence of a protease inhibitor. Cell lysate was immunoblotted for CD36 with β-actin used as a loading control. Results show that PD98059 did not block 7,8-NP’s effect on CD36 expression (Figure 3.4.3). 150µM 7,8-NP was found to significantly reduce CD36 expression to 41.20% of control, while 7,8-NP in conjunction with PD98059 reduced expression to a similar 48.66% of control. The differences between these two treatments were non-significant indicating that 7,8-NP did not down-regulate CD36 expression via the ERK 1/2 pathway. PD98059 treatment alone did not have a major effect on CED36 expression with a non-significant reduction to 75.17% of control, much less than was seen with other MAP kinase inhibitors.
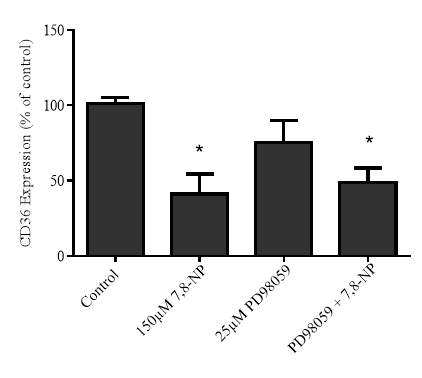
A
B

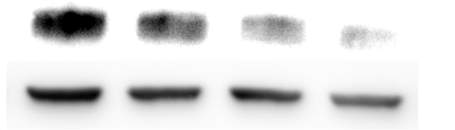
100 kDa
β-actin


Figure 3.4.3 MEK inhibition via PD98059 does not alleviate CD36 down regulation
U937 cells (0.5×106 cells/mL) were treated with 150µM 7,8-NP, 20µM of PD98059 and both in conjunction at 37oC over 24 hours in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of triplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots showed that SP600125 did not block the effect of 7,8-NP on CD36 expression to controls.
3.4.4 Effect of MEK inhibition on cellular viability
ERK1/2 cascades are involved in a wide variety of cellular functions including in adhesion, progression, migration, survival, differentiation, metabolism, proliferation, and transcription (Roskoski, 2012). Use of the PD98059 inhibitor would interfere with these processes with possible negative effects on cellular viability. To confirm the reliability of the results of the previous experiment cell viability was measured using the propidium iodide assay. Cells were incubated for 24 hours with either 25µM PD98059, 25µM PD98059 in conjunction with 150µM 7,8-NP, 150µM 7,8-NP alone or a cell only control. At the 24 hour time point cells were extracted and cell viability measured by PI using flow cytometry. Results show that at 25µM PD98059 had a limited effect on U937 cellular viability. After a 24 hour incubation viability had decreased to 0.67% lower than control (Figure 3.4.4). In contrast 150µM 7,8-NP viability was 1.33% higher than control and the two in combination 0.94% lower than control.
Changes in cell morphology after PD98059 treatment was examined using a light microscope (Leitz Wetzlab, Germany). Changes in cell morphology during treatment with PD98059 are non-existent with all cellular treatments showing a typical healthy morphology (Figure 3.4.4B).
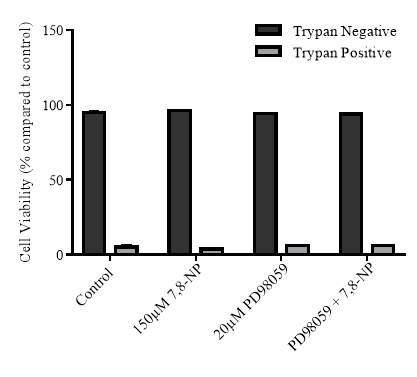
A
B


Control
150µM 7,8-NP


25µM PD98059
PD98059 + 7,8-NP
Figure 3.4.4 Effect of PD98059 on U937 cell viability using propidium iodide
U937 cells (0.5×106 cells/mL) were treated with 150µM 7,8-NP, 50µM of PD98059 and both in conjunction at 37oC over 24 hours in non-phenol red RPMI-1640. A) Cell viability was measured by PI and the data was expressed as a percentage of the respective control (cell only control). Results are displayed as mean ± SEM of triplicates from a single experiment, representative of three separate experiments. Significance levels are indicated as: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Images of U937 cells after treatment with 150µM 7,8-NP, 50µM of PD98059 and both in conjunction for 24 hours. Cells were viewed in tissue culture plates through an inverted microscope (20x magnification). The images were taken using a LEICA DMIL microscope with a LEICIA DFC290 camera.
3.4.5 Effect of p38 inhibition via SB202190 on 7,8-NP induced down regulation in the U937 cell line
Alongside the JNK and ERK1/2 kinases several other MAP kinases are involved in cell signalling and are candidates for involvement in 7,8-NP induced down regulation. The p38 MAP kinase and its signalling cascades are activated by stress and play a role in immune response, cellular survival and differentiation (Cuadrado & Nebreda, 2010). To explore the role of p38 in relation to 7,8-NP and CD36 the p38 inhibitor SB202190 was used. SB202190 is a selective, potent and cell permeable inhibitor of the p38 MAP kinase including both p38α and β isoforms. It works by binding within the ATP pocket of the active kinase preventing further signal transduction (S. P. Davies et al., 2000).
U937 cells where pre-incubated with 20µM SB202190 before 150µM 7,8-NP was added to selected wells to measure whether any additive effect between the two could be observed. 150µM 7,8-NP in isolation was used as a positive control and cells only as a negative control. At the 24 hour time point cells were extracted and lysed. Cell lysate was immunoblotted for CD36 with β-actin used as a loading control.
Results show that SB202190 did not block 7,8-NP’s effect on CD36. Rather like SP600125 it was able to decrease CD36 expression alone (Figure 3.4.5). This decrease was found to be 42.32% of control, slightly less than that of 7,8-NP which down regulated CD36 expression to 55.37% of control. When given in conjunction SB202190 and 7,8-NP lowered CD36 expression to 30.71% of control, which although is a larger decrease, is not significant compared to the two in isolation. All results were found to be significant in relation to control.
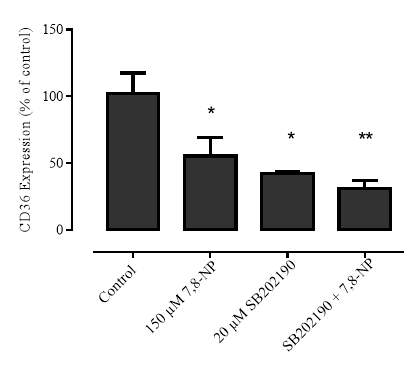
A
B


100 kDa
β-actin


Figure 3.4.5 p38 inhibition via SB202190 does not alleviate CD36 down regulation
U937 cells (0.5×106 cells/mL) were treated with 150µM 7,8-NP, 20µM of SB202190 and both in conjunction at 37oC over 24 hours in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of triplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots showed that SP600125 did not rescue the effect of 7,8-NP on CD36 expression relative to controls.
3.4.6 Effect of p38 inhibition on cellular viability
Due to the various roles that p38 plays in cellular signalling it is possible that the use of SB202190 would have an effect of cellular viability. To confirm the reliability of the results of the previous experiment cell viability was measured using the propidium iodide assay. Cells were incubated for 24 hours with either 20µM SB202190, 20µM SB202190 in conjunction with 150µM 7,8-NP, 150µM 7,8-NP alone or a cell only control. At the 24 hour time point cells were extracted and cell viability measured by propidium iodie using flow cytometry. Results show that 50µM SB202190 has a limited effect on U937 cellular viability. After a 24 hour incubation viability had non-significantly decreased to 2.02% lower than control (Figure 3.4.6). In contrast with the 150µM 7,8-NP treatment viability was 1.37% lower than control and with the two in combination viability was 2.26% lower than control.
Changes in cell morphology after GW992 treatment was examined using a light microscope (Leitz Wetzlab, Germany). Changes in cell morphology during treatment with SB202190 were limited with control cells showing a typical spherical morphology comparable to that seen with all other treatments (Figure 3.4.6).
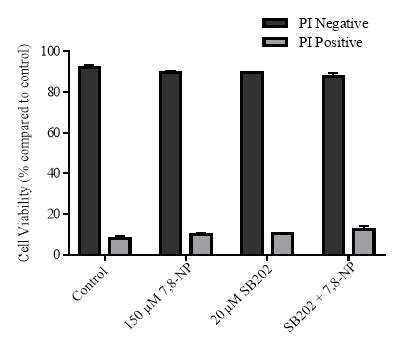
A
B


Control
150µM 7,8-NP


SB202190 + 7,8-NP
20µM SB202190
Figure 3.4.6 Effect of SB202190 on U937 cell viability using propidium iodide
U937 cells (0.5×106 cells/mL) were treated with 150µM 7,8-NP, 50µM of SB202190 and both in conjunction at 37oC over 24 hours in non-phenol red RPMI-1640. A) Cell viability was measured by PI and the data was expressed as a percentage of the respective control (cell only control). Results are displayed as mean ± SEM of triplicates from a single experiment, representative of three separate experiments. Significance levels are indicated as: (*) p ≤ 0.05, (**) p ≤ 0.01 and (**) p ≤ 0.001. B) Images of U937 cells after treatment with 150µM 7,8-NP, 50µM of SB202190 and both in conjunction for 24 hours. Cells were viewed in tissue culture plates through an inverted microscope (20x magnification). The images were taken using a LEICA DMIL microscope with a LEICIA DFC290 camera.
3.4.7 Effect of NF-kB inhibition via BAY 11-7082 on 7,8-NP induced down regulation in the U937 cell line
MAP kinases are not alone in the mediation of cellular signalling. The transcription factor NF-kB has been shown to be present in macrophages, and is involved in the transcription of numerous cytokines which play a role in cell growth and differentiation (Brasier, 2006). It was of interest here to explore the role of NF-kB inhibition on CD36 expression and whether this would have any effect on 7,8-NP induced down regulation. To achieve this U937 cells where pre-incubated with 2µM BAY 11-7082 before 150µM 7,8-NP was added to selected wells to measure whether any additive effect between the two could be observed. 150µM 7,8-NP in isolation was used as a positive control and cells only as a negative control. At the 24 hour time point cells were extracted and lysed in the presence of a protease inhibitor. Cell lysate was immunoblotted for CD36 with β-actin used as a loading control.
Results show that 2µM BAY 11-7082 did not alleviate 7,8-NP’s effect on CD36 expression (Figure 3.4.7). 150µM 7,8-NP was found to significantly reduce CD36 expression to 53.66% of control, while 7,8-NP in conjunction with 1µM BAY 11-7082 reduced expression to a similar 62.34% of control. The differences between these two treatments were non-significant indicating that 7,8-NP did not down-regulate CD36 expression via the NF-kB. 2µM BAY 11-7082 treatment alone was found to have an effect on CD36 expression with a significant reduction to 55.55% of control, similar to what was seen with the MAP kinase inhibitors.

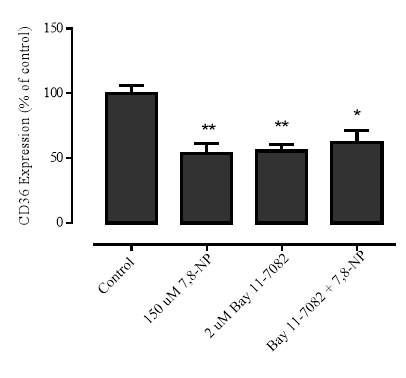

A
B
100 kDa
β-actin

150µM 7,8-NP
Figure 3.4.7 NF-kB inhibition via BAY 11-7082 does not alleviate CD36 down regulation
U937 cells (0.5×106 cells/mL) were treated with 150µM 7,8-NP, 2µM of BAY 11-7082 and both in conjunction at 37oC over 24 hours in RPMI-1640 with phenol red. Cell lysate was assessed for CD36 protein expression via western blot. A) Results were normalised by β-actin and displayed as mean ± SEM of triplicate experiments. Data is expressed as percentage of the respective control (cell only control) and significance is indicated from this control. Significance levels are: (*) p ≤ 0.05, (**) p ≤ 0.01 and (***) p ≤ 0.001. B) Quantitative densitometry of western blots showed that SP600125 did not rescue the effect of 7,8-NP on CD36 expression relative to controls.
3.4.8 Effect of NF-kB Inhibition on cellular viability
NF-kB plays an important role in the mediation of stress responses and inflammation. Use of the BAY 11-7082 inhibitor would interfere with these processes with possible negative effects on cellular viability. To confirm the reliability of the results of the previous experiment cell viability was measured using the propidium iodide assay. Cells were incubated for 24 hours with 2 µM BAY 11-7082, along with 2µM BAY 11-7082 in conjunction with 150µM 7,8-NP, 150µM 7,8-NP alone and a cell only control. At the 24 hour time point cells were extracted and cell viability measured by PI using flow cytometry under the FL-3 filter. Results show that 2µM BAY 11-7082 has a limited effect on U937 cellular viability after a 24 hour incubation in which viability had non-significantly decreased to 3.19% lower than control (Figure 3.4.8). In contrast with the 150µM 7,8-NP treatment viability was 1.37% lower than control and the two in combination 2.80% lower than control.
Changes in cell morphology after BAY 11-7082 treatment was examined using a light microscope (Leitz Wetzlab, Germany). Changes in cell morphology during treatment with BAY 11-7082 were limited with control cells showing a typical spherical morphology comparable to that seen with all other treatments (Figure 3.4.8).
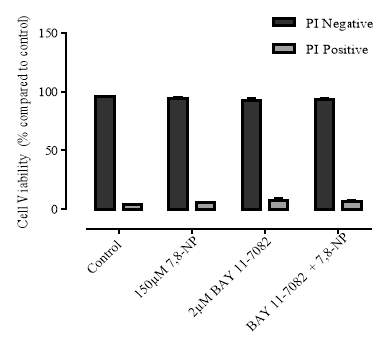
A
B

150µM 7,8-NP
Control


2µM BAY 11-7082
BAY 11-7082 + 7,8-NP
Figure 3.4.8 Effect of BAY 11-7082 on U937 cell viability using propidium iodide
U937 cells (0.5×106 cells/mL) were treated with 150µM 7,8-NP, 2 µM of BAY 11-7082 and both in conjunction at 37oC over 24 hours in non-phenol red RPMI-1640. A) Cell viability was measured by PI and the data was expressed as a percentage of the respective control (cell only control). Results are displayed as mean ± SEM of triplicates from a single experiment, representative of three separate experiments. Significance levels are indicated as: (*) p ≤ 0.05, (**) p ≤ 0.01 and (**) p ≤ 0.001. ) Images of U937 cells after treatment with 150µM 7,8-NP, 2µM of BAY 11-7082 and both in conjunction for 24 hours. Cells were viewed in tissue culture plates through an inverted microscope (20x magnification). The images were taken using a LEICA DMIL microscope with a LEICIA DFC290 camera.
3.5 CD36 flow cytometry
3.5.1 CD36 flow cytometry protocol design
Once CD36 down regulation had been characterised using western blotting the effect of 7,8-NP on CD36 surface expression in whole cells was examined using flow cytometry. Prior to this the effectiveness of the anti-CD36 antibody and PE labelled secondary antibody had to be re-determined after successful previous use in U937 cells and HMDMs (S. Davies, 2015). It was previously found that 10μg/mL of primary anti-CD36 antibody was the most effective concentration and this was re-examined here. U937 cells were treated in the dark, on ice with 10μg/mL of CD36 antibody, a primary, or a secondary antibody control for 30 minutes. Cells were suspended in 1mL PBS and BSA as per method and run through the flow cytometer using the FL2 filter which measured the fluorescence at 572 nM.
It was found the protocol used was reliable in detecting the CD36 bound antibody. Only one population was found to be present as expected when using an identical cell line (Figure 3.5.1.1). The presence of only one population means that any signal can be measured consistently due to the lack of interference from other cell populations. The second smaller cluster of cells is likely cellular debris. When gated in FL-2 all antibody controls emissions either had no (Cell only, 10) or very limited (20) overlap with the anti-CD36 signal. This indicates that 10μg/mL of primary antibody is an effective concentration to exclusively detect a strong CD36 signal. (Figure 3.5.1.2) shows a histogram along with analysis of results done in triplicate using GraphPad Prism 6.0.

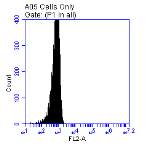
B
A
C
Cell Only Control

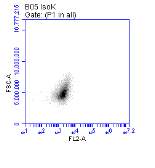
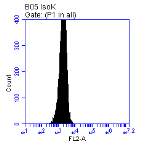

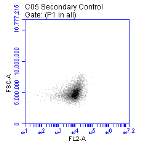
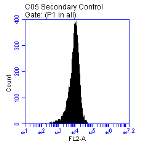

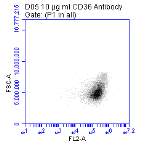
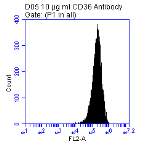
D
E
F
1o Control
G
H
I
2o Control
J
K
L
CD36 Antibody
Figure 3.5.1.1 Flow cytometer traces for CD36 antibody on U937 cells
U937 cells (0.5×106 cells/mL) were incubated with anti-CD36 primary and secondary antibodies as per method for 30 minutes before being processed by flow cytometry. Cell only, and 10 and 20 antibody controls were also used. A,D,G,J) indicates the presence of one dominant population of cells following incubation. E-F,H-I) show the position of FL-2 emission peak in the various controls. K & L) show that a strong anti CD36 signal was exclusively detected using 10µg/mL antibody.

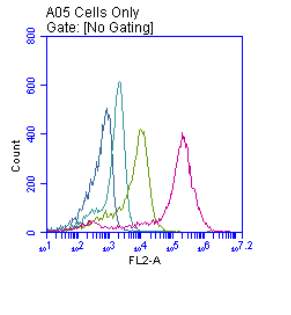
A
B
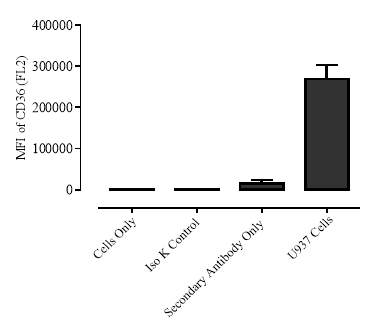
Figure 3.5.1.2 Use of the flow cytometry CD36 antibody on the U937 cell line
U937 cells (0.5×106 cells/mL) were incubated with anti-CD36 primary and secondary antibodies as per method for 30 minutes before being processed by flow cytometry. Cell only, and 10 and 20 antibody controls were also used. Whole cells were assessed for CD36 protein expression via flow cytometry A) FL-2 emission signal after treatment transformed into a histogram. B) Analysis of results done in triplicate using GraphPad Prism 6.0.
3.5.2 Down regulation of CD36 detected via flow cytometry
After confirmation of the flow cytometry protocol the effect of 7,8-NP on CD36 surface expression in whole cells was examined using flow cytometry. To achieve this U937 cells were incubated for 24 hours with 7,8-NP in concentrations ranging from 0-150µM. At the 24 hour time point the cells were extracted and treated in the dark, on ice with 10μg/mL of a CD36 antibody, a primary, or a secondary antibody control for 30 minutes. Cells were suspended in 1mL PBS and BSA as per method and run through the flow cytometer using the FL2 filter.
Results show that as the concentration of 7,8-NP increased the mean signal detected when gated in FL-2 decreased (Figures 3.5.2.1 & 3.5.2.2). At 50µM 7,8-NP mean FL-2 was 88.22% of control. The signal decreased further at 100µM (71.371%) and at 150µM (53.23%). The results indicate that 7,8-NP induced down regulation could be detected using flow cytometry, although not at the significance levels observed using western bolting. (Figure 3.5.2.1) also shows that as the mean FL-2 shifts towards the lower end of the spectrum a second population FL-2 gated cells can be seen. The shift does not correspond to second type of cell being present as only one population is still observed using forward and side scatter analysis. This indicates the difference in FL-2 gated emission comes from 7,8-NP having an effect on CD36 antibody binding to U937 cells due its presence and not an additional source. (Figure 3.5.2.2) shows a corresponding histogram of this shift along with analysis of results done in triplicate using GraphPad Prism 6.0.

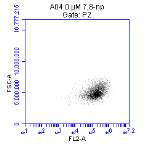
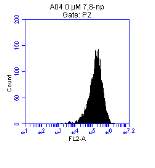
C
B
A
Cell Only Control

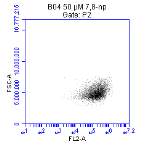
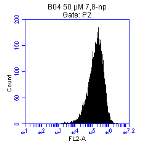
D
F
E
50 µM 7,8-NP
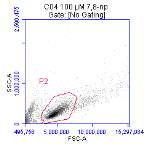
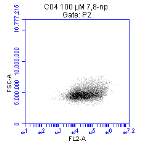
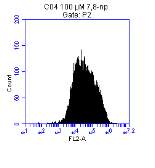
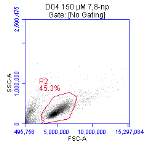
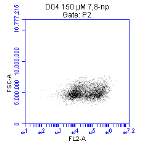
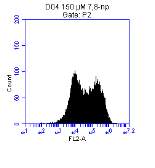
H
G
I
100 µM 7,8-NP
J
K
L
150 µM 7,8-NP
Figure 3.5.2.1 Flow cytometer traces for increasing concentrations of 7,8-NP on U937 cells
U937 cells (0.5×106 cells/mL) were treated with 0-150µM 7,8-NP and incubated at 37oC over a 24 hour time course. Whole cells were assessed for CD36 protein expression via flow cytometry. A,D,G,H) Indicates the presence of one dominant population of cells following incubation. B-C,E-F,H-I,K-L) Shows a decreasing emission signal at 572 nM as 7,8-NP concentration increases.

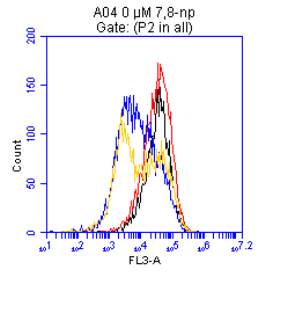
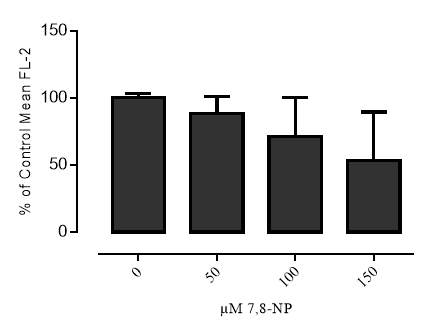
A
B
Figure 3.5.2.2 Effect of 7,8-NP on U937 cells detected using flow cytometry
U937 cells (0.5×106) were treated with 0-150 µM 7,8-NP and incubated at 37oC over a 24 hour time course. Whole cells were assessed for CD36 protein expression via flow cytometry A) FL-2 emission signal after treatment transformed into a histogram. B) Analysis of results done in triplicate using GraphPad Prism 6.0.
4. DISCUSSION
4.1 Characterising CD36 down regulation
It was found that in the U937 cell line 7,8-NP down regulated CD36 in a concentration dependant manner beginning at 50µM and continuing to 150µM (Figure 3.2.1). Noticeably the down regulation observed here was to a higher extent and occurred at lower concentrations compared to that previously seen when using Human Monocyte Derived Macrophages (HMDM) (S. Gieseg et al., 2010). This current research also explored the rate at which 7,8-NP had its down regulation effect on CD36 which begun at 6 hours and continued on to the 24 hour time point (Figure 3.2.2). Such rapid down-regulation was again not seen previously in HMDMs. These results suggest that 7,8-NP has a much stronger effect on U937 cells compared to HMDMs. A possible explanation for these discrepancies may be related to the inherent differences between monocytes and differentiated macrophages. Macrophages by nature are a lot bigger in size and it may be that the ratio of 7,8-NP to cell size has to be necessarily higher here in order to get the same result. It may also be that there are differences in the quantity of the 7,8-NP receptor between the cell types, and that it is differences in receptor-ligand binding that account for the increased effect seen in the U937 cell line. In the U937 cells 7,8-NP has been shown to enter the cell by the equilibrative nucleoside transporter 2 (ENT-2), whereas both ENT’s and concentrative nucleoside transporters (CNT) are involved in 7,8-NP uptake in human monocytes, which may correlate to macrophages (Janmale, 2013). Additionally it may be that the CD36 transcription is more sensitive to the effect of 7,8-NP in monocyte like cell systems.
Shchepetkina (2013) briefly also looked at the relationship between U937’s and 7,8-NP. However in comparison to the current research this relationship was not explored in depth. Schepetkina found previously that while the active band of CD36 was down regulated in the U937 cell line, the results were non-significant. A possible explanation for this discrepancy may be related to the basal intensities of CD36 in the cell batches used. In Shchepetkina’s research the CD36 band observed was much less compared to that seen in the present study, even though the same mature band was analysed. A result of this could be that a strong 7,8-NP effect may not have been possible to be detected. Evidence for this theory derives from one stage of the current research in which a fresh batch of U937 cells was brought up from storage that showed very little CD36 expression, and with 7,8-NP having no detectable effect. Upon examination it was found that this batch was much older compared to the prior batch used. It is possible that work the Shchepetkina (2013) undertook using the U937 cell line used a similar lower expressing batch which would explain the discrepancies in basal CD36 levels and the dampened effect of 7,8-NP.
To further characterise the effect of 7,8-NP it was of interest to explore if CD36 levels are able to recover after initial down regulation, and if so at what rate. This would allow for the use of the U937 cell line in treatments exploring the consequences of 7,8-NP down regulation along with examining any effects after recovery. Recovery was found to occur reasonably fast once 7,8-NP had been removed from the media with CD36 expression beginning to recover at 12 hours and nearing control levels 24 hours after removal (Figure 3.2.5). This result shows that future experimentation can be undertaken on U937 cells that have had their CD36 down regulated while still being viable. Of possible concern may be that full down regulation was not observed and that the rate of CD36 recovery limits the time frame for any experimentation to ensure CD36 levels are still down regulated. It is also of note that between the 24 and 48 hour time points CD36 expression began to decrease once again. This decrease did not correlated to loss of cellular viability (Figure 3.2.6). It is possible that the time course length by this point, totalling 48 hours, was having an impact and facilitating the cellular release of signals into the media that were impacting CD36 expression. Future experimentation will need to to examine what happens between the 24 and 48 hour time points. A possible remedy may be the replacement of cell media during recovery. At the point of writing it seems that this is the first research looking into the recovery of CD36 after down regulation limiting any comparison of these results.
In addition to examining the effect of 7,8-NP on CD36, the oxidised form of this pterin, neopterin, was also looked at (Figure 3.2.7). It was found that in the U937 cell line neopterin had no significant effect on CD36 expression. This result compares well to those of Shchepetkina (2013) who found that likewise neopterin had no significant effect on CD36 levels in HMDM cells. This suggests that a specific 7,8-NP property, and not a general pteridine effect, is likely responsible for CD36 down regulation. Yet other pteridines such as xanthopterin and biopterin were not examined during this research and it would be of further interest to examine their effect on CD36 to fully confirm this statement.
Overall the results here comprehensively show that 7,8-NP will down regulate CD36 in the U937 cell line, and has its effect at lower concentrations and at a faster rate compared to HMDMs. This supports their use in the current research exploring the relationship between 7,8-NP and CD36 with the aim of describing a possible mechanism of action.
4.2 CD36 Down Regulation Measured via Flow Cytometry
The western blotting protocol used in this thesis measured CD36 levels using cell lysate, specifically looking at the mature CD36 band located on the cell surface of U937s. The nature of western blotting required the cells to by fully lysed in order to separate out their proteins for antibody probing, consequently rendering the cells non-viable in the process. Flow cytometry was used as an alternative method to overcome this issue and enabled the anti-CD36 antibody to bind to the actual cell surface CD36 of whole cells.
Flow cytometery was considered an appropriate technique to use as changes in CD36 expression has previously been measured in monocyte cells using these protocols. Viana et al. (2005) showed a dose dependent increase in CD36 expression when THP-1’s were incubated in medium containing water soluble aldehydes and a significant reduction when incubated with α-tocopherol. While Carvalho et al. (2010), also using flow cytometry, demonstrated that HDL reduced measured CD36 in the U937 cell line when stimulated with oxLDL.
In the current research while a significant 7,8-NP induced CD36 down regulation event was observed using cell lysate, the same results were not seen when using flow cytometry. While a concentration depended down regulation was observed, the results after analysis were non-significant (Figure 3.5.1). This was not expected as the mature CD36 band analysed during the western blotting experiments is thought to correspond to cell surface CD36 (Alessio et al., 1996). One possible reason for this discrepancy in the results may be related to the recycling system a cell uses to replenish protein. There is a large amount of CD36 present in vacuoles that is not transported to the cell surface until required for protein turnover (Febbraio & Silverstein, 2007; Huh, Pearce, Yesner, Schindler, & Silverstein, 1996). It is possible that the differences in the effect seen may be related to this as vacuole contained protein would have been available after lysis for anti-body binding but not in whole cells.
Davies (2015) also using flow cytometry was able to measure the effect of 7,8-NP on CD36 using HMDMs. A small but significant reduction in CD36 was observed after a 24 hour incubation with 200µM 7,8-NP. Similar to U937s there were discrepancies between the flow cytometry results here and the effect that Shchepetkina (2013) found with HMDMs using western blotting. The combined results of this and the current work indicates that the overall sensitivity of the flow cytometry protocol is likely less compared to western blotting, and that if the viability of cells after analysis is not of concern, western blotting should be the favoured protocol to ensure a stronger measured effect.
4.3 Transcriptional Control of CD36
After confirming 7,8-NPs effect on CD36 in the U937 cell line the primary aim of this research was to describe the mechanism of its action. The first step here was to confirm what was under the transcriptional control of CD36 to ensure research into the appropriate system. While CD36 mRNA levels were not explored in this current research an impact on CD36 protein expression was thought likely to be the cause of 7,8-NP’s effect. This is due to research previously undertaken in the Free Radical Laboratory that showed that incubation with 7,8-NP significantly reduced the expression of CD36 mRNA in HMDM cells, pointing to an effect on CD36 transcriptional control (Shchepetkina, 2013).
Studies have shown that CD36 transcription is controlled by the heterodimer Peroxisome Proliferator Activated Receptor-γ/Retinoid X Receptor (PPARγ/RXR) (S. Han & Sidell, 2002; Tontonoz et al., 1998). This was confirmed in U937 cells via their incubation with the specific PPARγ inhibitor GW9662. Inhibition of PPARγ decreased in CD36 expression levels in a concentration dependant manner similar to what was seen after incubation with 7,8-NP (Figure 3.3.1).
This result correlated well to the experimental literature. Chen, Li, Wang, Wen, and Sun (2009) in research using differentiated L6 myotubes showed that incubation with GW9962 resulted in a decrease in basal CD36 levels to 64% of control. T.-S. Lee et al. (2009) found that resistin up-regulation of CD36 mediated by PPARγ was rescued by incubation with GW9662, and Flores et al. (2016)demonstrated that CD36 and PPARγ stimulated with the ligand 15d-PGJ2 was able to be inhibited by GW9662.
While it is the hypothesis of this thesis that 7,8-NP down regulates CD36 expression via interaction with the PPARɣ transcription factor, the use of the PPARɣ inhibitor did not necessarily confirm that 7,8-NP fully has its effect by preventing transcription. It is possible that 7,8-NP additionally has an effect down-stream of transcription and lowers CD36 levels by enhancing the degradation of this protein. Experiments with cycloheximide provided evidence that this is not the case. Cycloheximide produced by the bacterium Streptomyces griseus acts as an inhibitor of protein synthesis in Eukaryotic organisms by interfering with the translocation process (Jones & Cushman, 1989) meaning any protein loss subsequently observed is due to degradation of existing protein. A decrease in CD36 levels was found when U937 cells were incubated with both cycloheximide and cycloheximide in conjunction with 7,8-NP. Importantly down regulation under both treatments occurred at the same rate (Figure 3.3.5). It would have been expected that if 7,8-NP was having its effect by enhancing the degradation of CD36 that 7,8-NP incubation alongside cycloheximide would have had a much more significant impact on CD36 levels than incubation with cycloheximide alone. This not being the result provided strong evidence that 7,8-NP is indeed having its effect on CD36 during the transcriptional process. The results of the cycloheximide experiment additionally supplied information on the half-life of CD36 which was determined at being ~6 hours.
A variety of published research is available that also looked at the effect of cycloheximide on CD36 expression, but there is a wide discrepancy in the results that appears to be related to the cell line used. In J774A.1 macrophages 1µg/mL cycloheximide decreased CD36 expression to under 50% of control after 1 hour (Yun et al., 2008). This is a much faster rate than that observed in this research using U937 cells. Yet using a different type of macrophage (RAW264.7 macrophages) X. Yang et al. (2015) found a much slower rate using 5µM cycloheximide in which CD36 expression had decreased to ~60% of control after 16 hours. In human peripheral blood mononuclear cells, 1µg/mL cycloheximide diminished CD36 expression to around 50% of control after 48 hours (Malfitano et al., 2007), whileMwaikambo, Yang, Chemtob, and Hardy (2009) demonstrated that in human retinal pigment epithelial cells CD36 expression was found to be ~70% of control after 4 hours when incubated with 25µM cycloheximide. The large variability displayed between these results and what was found using the U937 cell line can possibly be explained by the inherent differences in the cell types used such as the size of the cell, differentiation state, the basal levels of CD36 present, along with the permeability of cycloheximide.
4.4 The Effect of MAP Kinase Inhibitors
The results of this research indicated that 7,8-NP is likely having its effect on CD36 via interfering with the PPARγ transcription factor. The regulatory process of PPARγ is complex with it containing both ligand and kinase binding sites which can either enhance or inhibit transcriptional activity (Adams et al., 1997; Feng et al., 2000; Hu et al., 1996). PPARγ also forms a heterodimer with RXR which itself can bind certain unsaturated fatty acids collectively termed rexnoids which have been shown to include docosahexaenoic acid, arachidonic acid and oleic acid (Dawson & Xia, 2012; Lengqvist et al., 2004). In terms of 7,8-NP’s mechanism of action it was thought that a MAP kinase mediated pathway was the most likely responsible for inhibiting CD36 transcription. It has been well established that the PPARγ kinase binding site recognises both the ERK1/2 and JNK kinases and that phosphorylation here is unusual in that it mediates a decrease in CD36 transcriptional activity (Adams et al., 1997; Hu et al., 1996). As such MAP kinases became the main focus of this thesis. Surprisingly it was found that inhibition of JNK and ERK1/2 kinases failed to rescue the effect of 7,8-NP, along with inhibition of additional p38 and NF-κB signalling pathways. While unexpectedly JNK, p38 and NF-κB inhibition alone down regulated CD36 levels.
4.4.1 JNK Inhibition
Evidence that JNK kinase mediated a decrease in PPARγ activity initially came from transfected cell studies. JNK kinase negatively regulated the transcriptional activity of human embryonic kidney cells transfected with mouse PPARγ via a PPARγ phosphorylation event (Camp & Tafuri, 1997), while a study by Adams et al. (1997) showed the same effect in JEG-3 cells transfected with human PPARγ.
It would have been expected that if 7,8-NP was having its effect on CD36 via activation of a JNK Kinase cascade that the use of the specific JNK inhibitor SP600125 would have blocked any down regulation. This was found not to be the case. While co-incubation of 7,8-NP with SP600125 lead to a slight rescue of CD36 expression, this was non-significant and importantly U937 incubation with SP600125 alone caused a large and significant decrease in CD36 (Figure 3.4.1). The viability experiments showed that the CD36 decrease was not the result of lost viability (Figure 3.4.2). Instead it appears that in model cell systems JNK mediated phosphorylation is not necessarily always responsible for a decrease in PPARγ transcriptional activity. Similar to the results found in this thesis, Huang et al. (2016)showed SP600125 treatment in the THP-1 cell line resulted in a decrease in basal levels of CD36, while also decreasing CD36 levels after oxLDL induced upregulation. A SP600125 mediated decrease in basal CD36 levels was also observed in research by Li, Kong, Li, He, and Zhou (2013) in THP-1 derived macrophages. Other cell systems have also found comparable results to what was observed using U937’s. In RAW264.7 macrophages JNK inhibition reduced the expression of CD36 in both cytosine-phosphate-guanine (CpG) stimulated and unstimulated cells. It was additionally found here that the phagocytic index decreased complementing the lowered CD36 effect (Wu et al., 2016). WhileRios, Gidlund, and Jancar (2011) showed that JNK inhibition reduced CD36 expression in adherent monocytes/macrophages stimulated with oxLDL.
These results alongside this current research seem to indicate that in certain cell lines such as U937s JNK is more responsible for the maintenance of basal CD36 levels and not for inducing a phosphorylation mediated down regulation event at PPARγ. This would explain as to why SP600125 is causing a reduction in CD36 levels when used alone, along with its failure to rescue 7,8-NP induced CD36 down regulation.
4.4.2 ERK1/2 Inhibition
The inefficiency of JNK inhibition to block 7,8-NP induced CD36 down regulation shifted the focus to the ERK1/2 kinase whose phosphorylation of PPARγ has also been demonstrated to decrease the transcription of CD36 (Adams et al., 1997; J. Han et al., 2000). To explore the role of ERK1/2 in this process, the MEK inhibitor (MAPKK that precedes ERK) PD98059 was used. The results here showed that similar to the effect seen with JNK inhibition, PD98059 failed to block 7,8-NP’s down regulation of CD36 (Figure 3.4.3). Yet while PPARγ phosphorylation by ERK1/2 has been shown to negatively impact CD36 transcription, the use of MEK inhibitors in other model monocyte/macrophage systems have additionally had contrasting results.
In experiments looking at THP-1 macrophages that had been stimulated with indoxyl suflate to take up oxLDL, upregulation of CD36, not down regulation, was inhibited via the use of MEK inhibitors (Cao, Fu, Wang, Jin, & Li, 2014). Cai, Lanting, and Natarajan (2004) again using THP-1 macrophages showed that upregulation of CD36 after binding to VSMCs was blocked by MEK inhibition in both bound and unbound THP-1s. WhileRios et al. (2011) using PBMC’s showed that upregulation of CD36 after stimulation with oxLDL could be reduced to basal levels via incubation with MEK inhibitors.
Similar to the results found when inhibiting JNK, the role of ERK1/2 appears to be more complex and is not only simply involved in the down regulation of CD36 transcription. Further research is required to fully identify the relationship between JNK and ERK1/2 kinase mediated phosphorylation and CD36 transcription in U937s, and to identify at what level of regulation they are having their strongest effect if not at PPARγ. Or whether if the phosphorylation mediated transcriptional decrease model is still viable when using other cell lines.
4.4.3 p38 Inhibition
The failure of inhibiting both kinases known to phosphorylate PPARγ in stopping 7,8-NP induced down regulation of CD36 shows that a MAP kinase cascade is unlikely to be the mechanism involved. Yet in carrying on with exploration of the kinase relationship to CD36, and due to the availability of its inhibitor, it was decided to additionally test the role of the common p38 kinase signalling pathway in this process. While p38 has not been demonstrated to have the ability to bind and phosphorylate PPARγ in vitro it was of interest to look at in this body of work due to it being role a major MAP kinase involved in cell signalling and with several publications having showed a relationship between p38 and PPARγ/CD36.
The results using the specific p38 inhibitor SB202190 showed a similar effect to that seen when inhibiting ERK1/2 and JNK, with p38 inhibition failing to block the down regulation effect 7,8-NP had on CD36 (Figure 3.4.3). It was interesting to note however that when the U937 cell line was solely incubated SB202190 a significant decrease in CD36 expression was observed. This type of result does not appear to be unique in the literature. Using primary trophoblasts Schild et al. (2006) demonstrated that p38 inhibitors decreased PPARγ’s transcriptional activity while having no effect on PPARγ expression. While research using 3T3-L1 fibroblasts showed that PPARγ activity is diminished in the presence of p38 inhibitors and additionally prevented lipid accumulation (Engelman, Lisanti, & Scherer, 1998), indicative of an effect of PPARγ target genes. Specifically in relation to CD36 Wu et al. (2016)showed that p38 inhibition reduced the expression of CD36 in both cytosine-phosphate-guanine (CpG) stimulated macrophages in which CD36 expression was enhanced, along with unstimulated cells. While M. Zhao et al. (2002) found that using a p38 inhibitor blocked foam cell formation in J774 cells exposed to oxLDL by reducing CD36 expression.
These results alongside the current research show that while p38 may not be phosphorylating PPARγ it is having a level of effect in relation to CD36, or could be interacting with PPARγ at an as yet unknown site. A possible explanation for this may be instead be related to PPARγ stabilization. Puigserver et al. (2001)identified a relationship between p38 and PPARγ in which cytokine activation of p38 resulted in the activation and stabilization of PPARγ coactivator-1. Prevention of this would necessarily decrease the ability of PPARγ to bind its DNA consensus sites and subsequently inhibit target gene transcription, including that of CD36.
4.4.4 NF-κB Inhibition
As shown in the experiments looking at JNK, ERK1/2 and p38 the use of MAP kinase inhibitors consistently failed to block 7,8-NP’s down regulation effect on CD36. It was next decided in carrying on with the theme of looking at cellular signalling to explore the effect of NF-κB inhibition on 7,8-NP induced down regulation due to the inhibitors being available in the Free Racial Biology Laboratory.
NF-κB has been shown to play a role in cellular inflammatory response and atherosclerosis (Pamukcu, Lip, & Shantsila, 2011). Under unstimulated conditions NF-κB exists in the cellular cytoplasm in an inhibited sate via interaction with iKB but can become activated upon stimulation with stress, cytokines, free radicals and bacterial antigens. After activation NF-κB disassociates with iKB and can translocate to the nucleus where it can induce the transcription of a number of genes including those involved in the production of cytokines, adhesion molecules chemokines and matrix metalloproteinases (Struzik, Szulc-Dabrowska, & Niemialtowski, 2014).
The results of this current research showed NF-κB inhibition via the specific inhibitor BAY 11-7082 failed to block 7,8-NP induced down regulation on CD36, and similar to JNK and p38, NF-κB inhibition itself was sufficient to decrease CD36 levels (Figure 3.4.7). The lack of an amelioratory effect was not overly surprising as NF-κB’s major role in lipid metabolism is related more to cholesterol efflux than influx (Tian & Tang, 2014; Xue et al., 2010; Yu et al., 2011; G.-j. Zhao et al., 2014), with the relationship between NF-κB and CD36 being more complex with contradictory experimental results. Masndose et al. showed that after Atorvastatin therapy, CD36 down regulation was associated with a significant reduction in the NF-κB levels of circulating monocytes in patients with Type II diabetes (Mandosi et al., 2010). Similarly a study showed that demethylated metabolites derived from nobiletin prevented NF-κB activity which was associated with reduced CD36 expression and cholestol uptake in the THP-1 cell line (Eguchi, Murakami, Li, Ho, & Ohigashi, 2007). Ferreira et al. (2007) et al. found an opposite effect in that macrophages with inactivated NF-κB were associated with an increase in CD36 mRNA and Ye, Jiang, Guo, Clark, and Gao (2013) showed that over expression of NF-κB p65 (a subunit of NF-κB transcription complex) resulted in reduced CD36 content and lipid content.
The results of the current experimental work aligns more with the first series of observations in which NF-κB inhibition was associated with a significant decrease in CD36 levels. This is similar to results found when using other monocytes but dissimilar to the macrophage experiments. It is possible that the differentiation state may have some level of impact on the effect of NF-κB in relation to CD36. Although importantly in relation to this thesis these results rule out a NF-κB mediated pathway for 7,8-NPs effect on CD36.
4.5 A different mechanism of effect
The inability of MAP kinase inhibitors to block 7,8-NPs effect on CD36 indicates that the down regulation is not occurring via a kinase pathway, or alternatively via the NF-κB pathway. This necessarily shifts the focus to a different mechanism of effect.
One possible pathway may involve a specific anti-oxidant activity of 7,8-NP. A wide variety of research has been published exploring the effects of antioxidants on CD36 using both cell and animal models. Using PMBC cells Dedoussis et al. (2004)demonstrated an effect with an antioxidant extract derived from Pistacia lentiscus. The extract restored Glutathione (GSH) levels, along with down-regulating CD36 expression including at the mRNA level in cells exposed to oxLDL. While using differentiated T3-L1 adipocytes it was found that the antioxidant molecules Oleuropein and Hydroxytyrosol exhibited a concentration dependent decrease in both PPARγ and CD36 levels (Drira, Chen, & Sakamoto, 2011). Treatment of SMCs and HL-60 macrophages with α-tocopherol down regulates CD36 expression (Ricciarelli et al., 2000). Paeonol, a potent antioxidant which has been shown to possess athero‑protective activity, significantly reduces intracellular lipid accumulation in macrophages along with inhibiting the mRNA and protein expression of CD36. In animal models Podszun et al. (2014)found that when Guinea Pigs on a high fat diet were supplemented with the antioxidant α-tocopherol or Atorvastatin CD36 levels were reduced to that of control. Likewise a study looking at the kidneys of rats undergoing chronic renal failure found that those supplemented with the antioxidant Niacin reduced the relative optical density of CD36 in renal tissue to lower than that of healthy controls (Cho, Kim, Kamanna, & Vaziri, 2010).
However a general antioxidant effect is unlikely to be responsible in terms of CD36 regulation as the down-regulation seen is not universal. Ricciarelli et al. (2000)who demonstrated the effect of α-tocopherol found no effect with either β-tocopherol or Procubul. While the antioxidant curcumin which lowered oxLDL induced cholesterol accumulation in macrophages, had no effect on CD36 (J. F. Zhao et al., 2012).
As a general anti-oxidant effect is unlikely, 7,8-NP needs to be able to act upon a specific pathway related to CD36 transcription. Alongside MAP kinase mediated phosphorylation PPARγ function can also be regulated by ligand binding. Of particular note is redox-regulation with several oxidised ligands having been shown to bind and activate PPARγ. The oxidised lipid 15-deoxy-Δ12,14-PGJ2 has been shown to act as a ligand (Forman et al., 1995),along with other polyunsaturated omega-6 fatty acid metabolites (Kliewer et al., 1997). 9- Hydroxyoctadecadienoic acid (9-HODE), 13-(HODE) along with 15(s)-HETE have also been shown to bind and promote PPARγ activation (Lenz et al., 1990; Nagy, Tontonoz, Alvarez, Chen, & Evans, 1998) Lipoxygenase (LPO) activity within the cell is responsible for the oxidation of phospholipids to HETES and HODES. As 7,8-NP is produced by the macrophage cell it is possible that 7,8-NP is inhibiting this LPO activity, thus stopping oxidised ligand formation and their interaction with PPARγ. This would explain the lack of effect of other antioxidants which may not necessarily be present in the appropriate location to inhibit this process.
7,8-NP may also be acting on a more specific pathway such as the JAK/STAT pathway. Activated STAT1 can form homodimers which when translocated to the nucleus will bind consensus DNA γ-activated sites which can lead to the transcription of CD36 (O’Shea et al., 2002; Ramana et al., 2002). Interaction of STAT1 with PPARγ requires it to become actytelated, but not phosphorylated. Acetylation requires p300 activation which in turn requires Syk2 and Pyk2 stimulation mediated by xanthine oxidase (XO) and importantly NADPH oxidise (NOX) ROS production. It is possible that 7,8-NPs mechanism of action involves radical scavenging of the ROS generated by NOX and XO and in so blocking STAT1 activation by this mechanism.
4.6 Incomplete Down regulation
It was of note that during this research that although down regulation of CD36 was able to be constantly observed and mediated via a range of conditions, complete removal of the CD36 band was not. The maximum average level of down regulation was consistently around ~40% of control. This result was not entirely unexpected as the processing of CD36 is such that it goes through several modification steps before it is able to be transferred from the Golgi apparatus to the cell surface. It is possible that the time allowed in the current experimentation was not enough to account for CD36 already synthesised but not yet fully transported. As shown in the cycloheximide experiment the half-life of detected CD36 was observed at being around 6 hours. This would indicate that the majority of the detected CD36 would have been ubiquintaed and degraded by the end of 24 hours. However immature CD36 would have been the least likely to be affected by this process and it is possible that the CD36 detected at the end of the down regulation experiments is a result of this residual CD36.
Another possible explanation for the lack of complete down regulation is the complexity of the PPARγ transcription system. PPARγ itself contains kinase and ligand binding sites, both of which are inter-related, while also forming a heterodimer with RXR which itself will bind ligands. It may be that 7,8-NP is only targeting one part of this system in an antagonistic way, while having limited interaction with the other components. This would allow for the continued interaction between the PPARγ:RXR heterodimer and additional agonists that compete with 7,8-NP induced down regulation, lessening its effect.
4.7 Future Research
Future research will need to explore additional mechanistic pathways to explain how 7,8-NP down regulates CD36. One obvious system would be to investigate the involvement of oxidised ligands and redox regulation in PPARγ activation and subsequent CD36 transcription. This would be able to be achieved by co-incubating U937 cells with synthetic PPARγ ligands alongside 7,8-NP. If 7,8-NP is having its effect via preventing the oxidation of fatty acids which then act as PPARγ ligands, the addition of further ligands at the right concentrations would be able to over come this effect. The role of ROS in STAT mediated CD36 regulation and the effect of 7,8-NP here should also be explored. Of particular note here would be the examination of the role that 15(s)-HETE plays in the activation of NADPH oxidase and whether 7,8-NP could prevent the subsequent ROS production leading to CD36 transcription. It would also be of interest to explore the mechanistic pathway of 7,8-NP induced down regulation in other cells lines apart from U937s, as the method of down regulation here would not necessarily be universal. The level of PPARγ and measurements of phosphorylated PPARγ should also be examined to fully confirm the kinase inhibitor studies
4.8 Summary
This research confirms that 7,8-NP down regulates CD36 in a time and concentration dependant manner in the U937 cell line. It was also shown that PPARγ is under transcriptional control of CD36 in the U937 cell line and that 7,8-NP is having its effect at the transcriptional level, and not by enhancing protein degradation. In relation to the mechanistic pathway, 7,8-NP is not down regulating CD36 via a MAP kinase cascade. Nor is it working via an NF-κB signalling pathway. Further research into the mechanism of 7,8-NP induced CD36 down regulation is required, including investigating the role that oxidised ligands plays in this process, and its effect on the STAT1 pathway.
BIBLIOGRAPHY
Abe, J., & Berk, B. C. (1998). Reactive oxygen species as mediators of signal transduction in cardiovascular disease. Trends in cardiovascular medicine, 8(2), 59-64.
Adams, M., Reginato, M. J., Shao, D., Lazar, M. A., & Chatterjee, V. K. (1997). Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. Journal of Biological Chemistry, 272(8), 5128-5132.
Agrawal, S., Febbraio, M., Podrez, E., Cathcart, M. K., Stark, G. R., & Chisolm, G. M. (2007). Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation, 115(23), 2939-2947.
Alessio, M., De Monte, L., Scirea, A., Gruarin, P., Tandon, N. N., & Sitia, R. (1996). Synthesis, processing, and intracellular transport of CD36 during monocytic differentiation. Journal of Biological Chemistry, 271(3), 1770-1775.
Armesilla, A. L., & Vega, M. A. (1994). Structural organization of the gene for human CD36 glycoprotein. Journal of Biological Chemistry, 269(29), 18985-18991.
Asch, A. S., Barnwell, J., Silverstein, R. L., & Nachman, R. L. (1987). Isolation of the thrombospondin membrane receptor. Journal of Clinical Investigation, 79(4), 1054.
Ashraf, M. Z., & Gupta, N. (2011). Scavenger receptors: implications in atherothrombotic disorders. The international journal of biochemistry & cell biology, 43(5), 697-700.
Asmis, R., & Jelk, J. (2000). Large variations in human foam cell formation in individuals: a fully autologous in vitro assay based on the quantitative analysis of cellular neutral lipids. Atherosclerosis, 148(2), 243-253.
Baird, S. K., Reid, L., Hampton, M. B., & Gieseg, S. P. (2005). OxLDL induced cell death is inhibited by the macrophage synthesised pterin, 7, 8-dihydroneopterin, in U937 cells but not THP-1 cells. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1745(3), 361-369.
Belkner, J., Stender, H., & Kühn, H. (1998). The rabbit 15-lipoxygenase preferentially oxygenates LDL cholesterol esters, and this reaction does not require vitamin E. Journal of Biological Chemistry, 273(36), 23225-23232.
Bennett, B. L., Sasaki, D. T., Murray, B. W., O’Leary, E. C., Sakata, S. T., Xu, W., . . . Satoh, Y. (2001). SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proceedings of the National Academy of Sciences, 98(24), 13681-13686.
Berliner, J. A., & Heinecke, J. W. (1996). The role of oxidized lipoproteins in atherogenesis. Free Radical Biology and Medicine, 20(5), 707-727.
Bobryshev, Y. V. (2006). Monocyte recruitment and foam cell formation in atherosclerosis. Micron, 37(3), 208-222.
Boström, P., Magnusson, B., Svensson, P.-A., Wiklund, O., Borén, J., Carlsson, L. M., . . . Hultén, L. M. (2006). Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arteriosclerosis, thrombosis, and vascular biology, 26(8), 1871-1876.
Boullier, A., Gillotte, K. L., Hörkkö, S., Green, S. R., Friedman, P., Dennis, E. A., . . . Quehenberger, O. (2000). The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of the lipoprotein. Journal of Biological Chemistry, 275(13), 9163-9169.
Brasier, A. R. (2006). The NF-κB regulatory network. Cardiovascular toxicology, 6(2), 111-130.
Brown, A. J., Dean, R. T., & Jessup, W. (1996). Free and esterified oxysterol: formation during copper-oxidation of low density lipoprotein and uptake by macrophages. Journal of lipid research, 37(2), 320-335.
Brown, A. J., Mander, E. L., Gelissen, I. C., Kritharides, L., Dean, R. T., & Jessup, W. (2000). Cholesterol and oxysterol metabolism and subcellular distribution in macrophage foam cells: accumulation of oxidized esters in lysosomes. Journal of lipid research, 41(2), 226-236.
Cai, Q., Lanting, L., & Natarajan, R. (2004). Interaction of monocytes with vascular smooth muscle cells regulates monocyte survival and differentiation through distinct pathways. Arteriosclerosis, thrombosis, and vascular biology, 24(12), 2263-2270.
Camp, H. S., & Tafuri, S. R. (1997). Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. Journal of Biological Chemistry, 272(16), 10811-10816.
Cao, L., Fu, Q., Wang, B. H., Jin, W., & Li, Z. (2014). Indoxyl sulfate stimulates oxidized LDL uptake through up-regulation of CD36 expression in THP-1 macrophages. Journal of Applied Biomedicine, 12(4), 203-209.
Carvalho, M. D. T., Vendrame, C. M. V., Ketelhuth, D. F. J., Yamashiro-Kanashiro, E. H., Goto, H., & Gidlund, M. (2010). High-density lipoprotein inhibits the uptake of modified low-density lipoprotein and the expression of CD36 and FcγRI. Journal of atherosclerosis and thrombosis, 17(8), 844-857.
Chanput, W., Peters, V., & Wichers, H. (2015). THP-1 and U937 cells The Impact of Food Bioactives on Health (pp. 147-159): Springer.
Chen, Y., Li, Y., Wang, Y., Wen, Y., & Sun, C. (2009). Berberine improves free-fatty-acid–induced insulin resistance in L6 myotubes through inhibiting peroxisome proliferator–activated receptor γ and fatty acid transferase expressions. Metabolism, 58(12), 1694-1702.
Cho, K.-h., Kim, H.-j., Kamanna, V. S., & Vaziri, N. D. (2010). Niacin improves renal lipid metabolism and slows progression in chronic kidney disease. Biochimica et Biophysica Acta (BBA)-General Subjects, 1800(1), 6-15.
Cobb, M. H., Boulton, T. G., & Robbins, D. J. (1991). Extracellular signal-regulated kinases: ERKs in progress. Cell regulation, 2(12), 965.
Collot-Teixeira, S., Martin, J., McDermott-Roe, C., Poston, R., & McGregor, J. L. (2007). CD36 and macrophages in atherosclerosis. Cardiovascular research, 75(3), 468-477.
Cuadrado, A., & Nebreda, A. R. (2010). Mechanisms and functions of p38 MAPK signalling. Biochemical Journal, 429(3), 403-417.
D’Archivio, M., Scazzocchio, B., Filesi, C., Varì, R., Maggiorella, M. T., Sernicola, L., . . . Masella, R. (2008). Oxidised LDL up‐regulate CD36 expression by the Nrf2 pathway in 3T3‐L1 preadipocytes. FEBS letters, 582(15), 2291-2298.
Dahlöf, B. (2010). Cardiovascular disease risk factors: epidemiology and risk assessment. The American journal of cardiology, 105(1), 3A-9A.
Dántola, M. L., Vignoni, M., Capparelli, A. L., Lorente, C., & Thomas, A. H. (2008). Stability of 7, 8‐Dihydropterins in air‐equilibrated aqueous solutions. Helvetica Chimica Acta, 91(3), 411-425.
Davies, C., & Tournier, C. (2012). Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies: Portland Press Limited.
Davies, S. (2015). 7,8-Dihydroneopterin and its effect on the formation of foam cells.
Davies, S. P., Reddy, H., Caivano, M., & Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochemical Journal, 351(1), 95-105.
Dawson, M. I., & Xia, Z. (2012). The retinoid X receptors and their ligands. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids, 1821(1), 21-56.
Dedoussis, G. V., Kaliora, A. C., Psarras, S., Chiou, A., Mylona, A., Papadopoulos, N. G., & Andrikopoulos, N. K. (2004). Antiatherogenic effect of Pistacia lentiscus via GSH restoration and downregulation of CD36 mRNA expression. Atherosclerosis, 174(2), 293-303.
Doi, T., Higashino, K.-I., Kurihara, Y., Wada, Y., Miyazaki, T., Nakamura, H., . . . Itakura, H. (1993). Charged collagen structure mediates the recognition of negatively charged macromolecules by macrophage scavenger receptors. Journal of Biological Chemistry, 268(3), 2126-2133.
Draude, G., & Lorenz, R. L. (2000). TGF-β1 downregulates CD36 and scavenger receptor A but upregulates LOX-1 in human macrophages. American Journal of Physiology-Heart and Circulatory Physiology, 278(4), H1042-H1048.
Dreyer, C., Krey, G., Keller, H., Givel, F., Helftenbein, G., & Wahli, W. (1992). Control of the peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell, 68(5), 879-887.
Drira, R., Chen, S., & Sakamoto, K. (2011). Oleuropein and hydroxytyrosol inhibit adipocyte differentiation in 3 T3-L1 cells. Life sciences, 89(19), 708-716.
Duggan, S., Rait, C., Gebicki, J. M., & Gieseg, S. (2001). Inhibition of protein oxidation by the macrophage-synthesised antioxidant 7, 8-dihydroneopterin. Redox Report, 6(3), 188-190.
Duggan, S., Rait, C., Platt, A., & Gieseg, S. P. (2002). Protein and thiol oxidation in cells exposed to peroxyl radicals is inhibited by the macrophage synthesised pterin 7, 8-dihydroneopterin. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1591(1), 139-145.
Eguchi, A., Murakami, A., Li, S., Ho, C.-T., & Ohigashi, H. (2007). Suppressive effects of demethylated metabolites of nobiletin on phorbol ester-induced expression of scavenger receptor genes in THP-1 human monocytic cells. Biofactors, 31(2), 107-116.
Endemann, G., Stanton, L., Madden, K., Bryant, C., White, R. T., & Protter, A. (1993). CD36 is a receptor for oxidized low density lipoprotein. Journal of Biological Chemistry, 268(16), 11811-11816.
Engelman, J. A., Lisanti, M. P., & Scherer, P. E. (1998). Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. Journal of Biological Chemistry, 273(48), 32111-32120.
Esterbauer, H., Dieber-Rotheneder, M., Waeg, G., Striegl, G., & Juergens, G. (1990). Biochemical structural and functional properties of oxidized low-density lipoprotein. Chemical research in toxicology, 3(2), 77-92.
Evanko, S. P., Raines, E. W., Ross, R., Gold, L. I., & Wight, T. N. (1998). Proteoglycan distribution in lesions of atherosclerosis depends on lesion severity, structural characteristics, and the proximity of platelet-derived growth factor and transforming growth factor-beta. The American journal of pathology, 152(2), 533.
Falk, E. (1983). Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. British heart journal, 50(2), 127-134.
Febbraio, M., Podrez, E. A., Smith, J. D., Hajjar, D. P., Hazen, S. L., Hoff, H. F., . . . Silverstein, R. L. (2000). Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. The Journal of clinical investigation, 105(8), 1049-1056.
Febbraio, M., & Silverstein, R. L. (2007). CD36: implications in cardiovascular disease. The international journal of biochemistry & cell biology, 39(11), 2012-2030.
Feng, J., Han, J., Pearce, S. F. A., Silverstein, R. L., Gotto, A. M., Hajjar, D. P., & Nicholson, A. C. (2000). Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-γ. Journal of lipid research, 41(5), 688-696.
Fernández-Ruiz, E., Armesilla, A. L., Sánchez-Madrid, F., & Vega, M. A. (1993). Gene encoding the collagen type I and thrombospondin receptor CD36 is located on chromosome 7q11. 2. Genomics, 17(3), 759-761.
Ferreira, V., van Dijk, K. W., Groen, A. K., Vos, R. M., van der Kaa, J., Gijbels, M. J., . . . Pannekoek, H. (2007). Macrophage-specific inhibition of NF-κB activation reduces foam-cell formation. Atherosclerosis, 192(2), 283-290.
Firth, C. A., Crone, E. M., Flavall, E. A., Roake, J. A., & Gieseg, S. P. (2008). Macrophage mediated protein hydroperoxide formation and lipid oxidation in low density lipoprotein are inhibited by the inflammation marker 7, 8-dihydroneopterin. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1783(6), 1095-1101.
Flores, J. J., Klebe, D., Rolland, W. B., Lekic, T., Krafft, P. R., & Zhang, J. H. (2016). PPARγ-induced upregulation of CD36 enhances hematoma resolution and attenuates long-term neurological deficits after germinal matrix hemorrhage in neonatal rats. Neurobiology of disease, 87, 124-133.
Forman, B. M., Tontonoz, P., Chen, J., Brun, R. P., Spiegelman, B. M., & Evans, R. M. (1995). 15-deoxy-Δ12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell, 83(5), 803-812.
Fuchs, D., Avanzas, P., Arroyo-Espliguero, R., Jenny, M., Consuegra-Sanchez, L., & Kaski, J. (2009). The role of neopterin in atherogenesis and cardiovascular risk assessment. Current medicinal chemistry, 16(35), 4644-4653.
Galis, Z. S., Sukhova, G. K., Lark, M. W., & Libby, P. (1994). Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. Journal of Clinical Investigation, 94(6), 2493.
Gao, B. (2005). Cytokines, STATs and liver disease. Cell Mol Immunol, 2(2), 92-100.
Geng, Y.-j., Holm, J., Nygren, S., Bruzelius, M., Stemme, S., & Hansson, G. K. (1995). Expression of the macrophage scavenger receptor in atheroma. Arteriosclerosis, thrombosis, and vascular biology, 15(11).
Gieseg, S., Amit, Z., Yang, Y.-T., Shchepetkina, A., & Katouah, H. (2010). Oxidant production, oxLDL uptake, and CD36 levels in human monocyte–derived macrophages are downregulated by the macrophage-generated antioxidant 7, 8-dihydroneopterin. Antioxidants & redox signaling, 13(10), 1525-1534.
Gieseg, S., & Cato, S. (2003). Inhibition of THP-1 cell-mediated low-density lipoprotein oxidation by the macrophage-synthesised pterin, 7, 8-dihydroneopterin. Redox report, 8(2), 113-115.
Gieseg, S., Crone, E., Flavall, E., & Amit, Z. (2008). Potential to inhibit growth of atherosclerotic plaque development through modulation of macrophage neopterin/7, 8‐dihydroneopterin synthesis. British journal of pharmacology, 153(4), 627-635.
Gieseg, S., Leake, D. S., Flavall, E. M., Amit, Z., Reid, L., & Yang, Y.-T. (2008). Macrophage antioxidant protection within atherosclerotic plaques. Frontiers in bioscience (Landmark edition), 14, 1230-1246.
Gieseg, S., Maghzal, G., & Glubb, D. (2001). Protection of erythrocytes by the macrophage synthesized antioxidant 7, 8 dihydroneopterin. Free radical research, 34(2), 123-136.
Gieseg, S., Reibnegger, G., Wachter, H., & Esterbauer, H. (1995). 7, 8 Dihydroneopterin inhibits low density lipoprotein oxidation in vitro. Evidence that this macrophage secreted pteridine is an anti-oxidant. Free radical research, 23(2), 123-136.
Gieseg, S. P., Amit, Z., Yang, Y.-T., Shchepetkina, A., & Katouah, H. (2010). Oxidant production, oxLDL uptake, and CD36 levels in human monocyte–derived macrophages are downregulated by the macrophage-generated antioxidant 7, 8-dihydroneopterin. Antioxidants & redox signaling, 13(10), 1525-1534.
Greenwalt, D. E., Lipsky, R. H., Ockenhouse, C. F., Ikeda, H., Tandon, N. N., & Jamieson, G. (1992). Membrane glycoprotein CD36: a review of its roles in adherence, signal transduction, and transfusion medicine. Blood, 80(5), 1105-1115.
Griendling, K. K., Sorescu, D., & Ushio-Fukai, M. (2000). Nad (p) h Oxidase. Circulation research, 86(5), 494-501.
Han, J., Hajjar, D. P., Tauras, J. M., Feng, J., Gotto, A. M., & Nicholson, A. C. (2000). Transforming growth factor-β1 (TGF-β1) and TGF-β2 decrease expression of CD36, the type B scavenger receptor, through mitogen-activated protein kinase phosphorylation of peroxisome proliferator-activated receptor-γ. Journal of Biological Chemistry, 275(2), 1241-1246.
Han, S., & Sidell, N. (2002). Peroxisome‐proliferator‐activated‐receptor gamma (PPARγ) independent induction of CD36 in THP‐1 monocytes by retinoic acid. Immunology, 106(1), 53-59.
Hansson, G. K. (2001). Immune mechanisms in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology, 21(12), 1876-1890.
Hansson, G. K. (2005). Inflammation, atherosclerosis, and coronary artery disease. New England Journal of Medicine, 352(16), 1685-1695.
Hansson, G. K., & Hermansson, A. (2011). The immune system in atherosclerosis. Nature immunology, 12(3), 204-212.
Hardwick, J. P., Osei-Hyiaman, D., Wiland, H., Abdelmegeed, M. A., & Song, B.-J. (2010). PPAR/RXR Regulation of Fatty Acid Metabolism and Fatty Acid PPAR research, 2009.
Henriksen, T., Mahoney, E. M., & Steinberg, D. (1983). Enhanced macrophage degradation of biologically modified low density lipoprotein. Arteriosclerosis, Thrombosis, and Vascular Biology, 3(2), 149-159.
Ho, Y., Brown, S., Bilheimer, D. W., & Goldstein, J. L. (1976). Regulation of low density lipoprotein receptor activity in freshly isolated human lymphocytes. Journal of Clinical Investigation, 58(6), 1465.
Hu, E., Kim, J. B., Sarraf, P., & Spiegelman, B. M. (1996). Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPAR gamma. Science, 274(5295), 2100.
Huang, E.-W., Liu, C.-Z., Liang, S.-J., Zhang, Z., Lv, X.-F., Liu, J., . . . Guan, Y.-Y. (2016). Endophilin-A2-mediated increase in scavenger receptor expression contributes to macrophage-derived foam cell formation. Atherosclerosis, 254, 133-141.
Huber, C., Batchelor, J. R., Fuchs, D., Hausen, A., Lang, A., Niederwieser, D., . . . Wachter, H. (1984). Immune response-associated production of neopterin. Release from macrophages primarily under control of interferon-gamma. The Journal of experimental medicine, 160(1), 310-316.
Huh, H., Pearce, S., Yesner, L., Schindler, J., & Silverstein, R. (1996). Regulated expression of CD36 during monocyte-to-macrophage differentiation: potential role of CD36 in foam cell formation. Blood, 87(5), 2020-2028.
Ishida, M., Ishida, T., Thomas, S. M., & Berk, B. C. (1998). Activation of extracellular signal–regulated kinases (ERK1/2) by angiotensin II is dependent on c-Src in vascular smooth muscle cells. Circulation research, 82(1), 7-12.
Janmale, T. (2013). Formation, Transport and Detection of 7,8-Dihydroneopterin. University of Canterbury.
Janmale, T., Genet, R., Crone, E., Flavall, E., Firth, C., Pirker, J., . . . Gieseg, S. P. (2015). Neopterin and 7, 8-dihydroneopterin are generated within atherosclerotic plaques. Pteridines, 26(3), 93-103.
Jessup, W., & Kritharides, L. (2000). Metabolism of oxidized LDL by macrophages. Current opinion in lipidology, 11(5), 473-481.
Johnson, W. J., Mahlberg, F. H., Rothblat, G. H., & Phillips, M. C. (1991). Cholesterol transport between cells and high-density lipoproteins. Biochimica et Biophysica Acta (BBA)-Lipids and Lipid Metabolism, 1085(3), 273-298.
Jones, T., & Cushman, S. W. (1989). Acute effects of cycloheximide on the translocation of glucose transporters in rat adipose cells. Journal of Biological Chemistry, 264(14), 7874-7877.
Katouah, H., Chen, A., Othman, I., & Gieseg, S. P. (2015). Oxidised low density lipoprotein causes human macrophage cell death through oxidant generation and inhibition of key catabolic enzymes. The international journal of biochemistry & cell biology, 67, 34-42.
Kliewer, S. A., Sundseth, S. S., Jones, S. A., Brown, P. J., Wisely, G. B., Koble, C. S., . . . Lenhard, J. M. (1997). Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proceedings of the National Academy of Sciences, 94(9), 4318-4323.
Kodama, T., Reddy, P., Kishimoto, C., & Krieger, M. (1988). Purification and characterization of a bovine acetyl low density lipoprotein receptor. Proceedings of the National Academy of Sciences, 85(23), 9238-9242.
Kunjathoor, V. V., Febbraio, M., Podrez, E. A., Moore, K. J., Andersson, L., Koehn, S., . . . Freeman, M. W. (2002). Scavenger receptors class AI/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. Journal of Biological Chemistry, 277(51), 49982-49988.
Lee, S., Pineau, T., Drago, J., Lee, E. J., Owens, J. W., Kroetz, D. L., . . . Gonzalez, F. J. (1995). Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Molecular and cellular biology, 15(6), 3012-3022.
Lee, T.-S., Lin, C.-Y., Tsai, J.-Y., Wu, Y.-L., Su, K.-H., Lu, K.-Y., . . . Hsu, Y.-P. (2009). Resistin increases lipid accumulation by affecting class A scavenger receptor, CD36 and ATP-binding cassette transporter-A1 in macrophages. Life sciences, 84(3), 97-104.
Leesnitzer, L. M., Parks, D. J., Bledsoe, R. K., Cobb, J. E., Collins, J. L., Consler, T. G., . . . Patel, L. (2002). Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry, 41(21), 6640-6650.
Lengqvist, J., de Urquiza, A. M., Bergman, A.-C., Willson, T. M., Sjövall, J., Perlmann, T., & Griffiths, W. J. (2004). Polyunsaturated fatty acids including docosahexaenoic and arachidonic acid bind to the retinoid X receptor α ligand-binding domain. Molecular & Cellular Proteomics, 3(7), 692-703.
Lenz, M., Hughes, H., Mitchell, J., Via, D., Guyton, J., Taylor, A., . . . Smith, C. V. (1990). Lipid hydroperoxy and hydroxy derivatives in copper-catalyzed oxidation of low density lipoprotein. Journal of lipid research, 31(6), 1043-1050.
Leon, M. A., & Zuckerman, S. (2005). Gamma interferon: a central mediator in atherosclerosis. Inflammation Research, 54(10), 395-411.
Levitan, I., Volkov, S., & Subbaiah, P. V. (2010). Oxidized LDL: diversity, patterns of recognition, and pathophysiology. Antioxidants & redox signaling, 13(1), 39-75.
Lewis, B. (1973). Classification of lipoproteins and lipoprotein disorders. Journal of Clinical Pathology. Supplement (Ass. Clin. Path.), 5, 26.
Li, X., Kong, L.-X., Li, J., He, H.-X., & Zhou, Y.-D. (2013). Kaempferol suppresses lipid accumulation in macrophages through the downregulation of cluster of differentiation 36 and the upregulation of scavenger receptor class B type I and ATP-binding cassette transporters A1 and G1. International journal of molecular medicine, 31(2), 331-338.
Libby, P., & Aikawa, M. (1998). New insights into plaque stabilisation by lipid lowering. Drugs, 56(1), 9-13.
Libby, P., Ridker, P. M., & Hansson, G. K. (2009). Inflammation in atherosclerosis: from pathophysiology to practice. Journal of the American College of Cardiology, 54(23), 2129-2138.
Libby, P., Ridker, P. M., & Maseri, A. (2002). Inflammation and atherosclerosis. Circulation, 105(9), 1135-1143.
Liu, Z., & Li, Y. (2013). Relationship between serum neopterin levels and coronary heart disease. Genet. Mol. Res, 12(4), 4222-4229.
Lusis, A. J. (2000). Atherosclerosis. Nature, 407(6801), 233-241. doi: 10.1038/35025203
Mach, F., Schönbeck, U., Bonnefoy, J.-Y., Pober, J. S., & Libby, P. (1997). Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40. Circulation, 96(2), 396-399.
Madamanchi, N. R., Vendrov, A., & Runge, M. S. (2005). Oxidative stress and vascular disease. Arteriosclerosis, thrombosis, and vascular biology, 25(1), 29-38.
Malek, A. M., Alper, S. L., & Izumo, S. (1999). Hemodynamic shear stress and its role in atherosclerosis. Jama, 282(21), 2035-2042.
Malfitano, A. M., Toruner, G. A., Gazzerro, P., Laezza, C., Husain, S., Eletto, D., . . . Schwalb, M. (2007). Arvanil and anandamide up-regulate CD36 expression in human peripheral blood mononuclear cells. Immunology letters, 109(2), 145-154.
Mandosi, E., Fallarino, M., Gatti, A., Carnovale, A., Rossetti, M., Lococo, E., . . . Morano, S. (2010). Atorvastatin downregulates monocyte CD36 expression, nuclear NFkappaB and TNFalpha levels in type 2 diabetes. J Atheroscler Thromb, 17(6), 539-545.
Maor, I., & Aviram, M. (1994). Oxidized low density lipoprotein leads to macrophage accumulation of unesterified cholesterol as a result of lysosomal trapping of the lipoprotein hydrolyzed cholesteryl ester. Journal of lipid research, 35(5), 803-819.
Matsumoto, K., Hirano, K., Nozaki, S., Takamoto, A., Nishida, M., Nakagawa-Toyama, Y., . . . Matsuzawa, Y. (2000). Expression of macrophage (Mφ) scavenger receptor, CD36, in cultured human aortic smooth muscle cells in association with expression of peroxisome proliferator activated receptor-γ, which regulates gain of Mφ-like phenotype in vitro, and its implication in atherogenesis. Arteriosclerosis, thrombosis, and vascular biology, 20(4), 1027-1032.
McGregor, J., Catimel, B., Parmentier, S., Clezardin, P., Dechavanne, M., & Leung, L. (1989). Rapid purification and partial characterization of human platelet glycoprotein IIIb. Interaction with thrombospondin and its role in platelet aggregation. Journal of Biological Chemistry, 264(1), 501-506.
Meyer, J. W., & Schmitt, M. E. (2000). A central role for the endothelial NADPH oxidase in atherosclerosis. FEBS letters, 472(1), 1-4.
Moore, K. J., & Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell, 145(3), 341-355.
Mosser, D. M., & Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nature reviews immunology, 8(12), 958-969.
Munteanu, A., Taddei, M., Tamburini, I., Bergamini, E., Azzi, A., & Zingg, J.-M. (2006). Antagonistic effects of oxidized low density lipoprotein and α-tocopherol on CD36 scavenger receptor expression in monocytes involvment of protein kinase and peroxisome proliferator-activated receptor-γ. Journal of Biological Chemistry, 281(10), 6489-6497.
Murr, C., Widner, B., Wirleitner, B., & Fuchs, D. (2002). Neopterin as a marker for immune system activation. Current drug metabolism, 3(2), 175-187.
Mwaikambo, B. R., Yang, C., Chemtob, S., & Hardy, P. (2009). Hypoxia up-regulates CD36 expression and function via hypoxia-inducible factor-1-and phosphatidylinositol 3-kinase-dependent mechanisms. Journal of Biological Chemistry, 284(39), 26695-26707.
Nagy, L., Tontonoz, P., Alvarez, J. G., Chen, H., & Evans, R. M. (1998). Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell, 93(2), 229-240.
Nakashima, Y., Wight, T. N., & Sueishi, K. (2008). Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovascular research, 79(1), 14-23.
Navazo, M. D. P., Daviet, L., Ninio, E., & McGregor, J. L. (1996). Identification on human CD36 of a domain (155-183) implicated in binding oxidized low-density lipoproteins (Ox-LDL). Arteriosclerosis, thrombosis, and vascular biology, 16(8), 1033-1039.
Nicholson, A. C., Frieda, S., Pearce, A., & Silverstein, R. L. (1995). Oxidized LDL Binds to CD36 on Human Monocyte-Derived Macrophages and Transfected Cell Lines Evidence Implicating the Lipid Moiety of the Lipoprotein as the Binding Site. Arteriosclerosis, thrombosis, and vascular biology, 15(2), 269-275.
Nozaki, S., Kashiwagi, H., Yamashita, S., Nakagawa, T., Kostner, B., Tomiyama, Y., . . . Kameda-Takemura, K. (1995). Reduced uptake of oxidized low density lipoproteins in monocyte-derived macrophages from CD36-deficient subjects. Journal of Clinical Investigation, 96(4), 1859.
O’Shea, J. J., Gadina, M., & Schreiber, R. D. (2002). Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell, 109(2), S121-S131.
Obama, T., Kato, R., Masuda, Y., Takahashi, K., Aiuchi, T., & Itabe, H. (2007). Analysis of modified apolipoprotein B‐100 structures formed in oxidized low‐density lipoprotein using LC‐MS/MS. Proteomics, 7(13), 2132-2141.
Oettl, K., Greilberger, J., Dikalov, S., & Reibnegger, G. (2004). Interference of 7,8-dihydroneopterin with peroxynitrite-mediated reactions. Biochemical and biophysical research communications, 321(2), 379-385.
Oettl, K., & Reibnegger, G. (2002). Pteridine derivatives as modulators of oxidative stress. Current Drug Metabolism, 3(2), 203-209.
Ohgami, N., Nagai, R., Ikemoto, M., Arai, H., Kuniyasu, A., Horiuchi, S., & Nakayama, H. (2001). CD36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. Journal of Biological Chemistry, 276(5), 3195-3202.
Ohidar, R. S. (2010). Mechanisms of cell signaling by the scavenger receptor CD36: implications in atherosclerosis and thrombosis. Transactions of the American Clinical and Climatological Association, 121.
Ono, K., & Han, J. (2000). The p38 signal transduction pathway activation and function. Cellular signalling, 12(1), 1-13.
Ozcan, L., & Tabas, I. (2016). Calcium signalling and ER stress in insulin resistance and atherosclerosis. Journal of Internal Medicine, 280(5), 457-464.
Pamukcu, B., Lip, G. Y., & Shantsila, E. (2011). The nuclear factor–kappa B pathway in atherosclerosis: a potential therapeutic target for atherothrombotic vascular disease. Thrombosis research, 128(2), 117-123.
Park, Y. M., Febbraio, M., & Silverstein, R. L. (2009). CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. The Journal of clinical investigation, 119(1), 136-145.
Pearce, S. F. A., Roy, P., Nicholson, A. C., Hajjar, D. P., Febbraio, M., & Silverstein, R. L. (1998). Recombinant glutathione S-transferase/CD36 fusion proteins define an oxidized low density lipoprotein-binding domain. Journal of Biological Chemistry, 273(52), 34875-34881.
Podrez, E. A., Poliakov, E., Shen, Z., Zhang, R., Deng, Y., Sun, M., . . . Fox, P. L. (2002). Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. Journal of Biological Chemistry, 277(41), 38503-38516.
Podszun, M. C., Grebenstein, N., Spruss, A., Schlueter, T., Kremoser, C., Bergheim, I., & Frank, J. (2014). Dietary α-tocopherol and atorvastatin reduce high-fat-induced lipid accumulation and down-regulate CD36 protein in the liver of guinea pigs. The Journal of nutritional biochemistry, 25(5), 573-579.
Potteaux, S., Gautier, E. L., Hutchison, S. B., van Rooijen, N., Rader, D. J., Thomas, M. J., . . . Randolph, G. J. (2011). Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe–/–mice during disease regression. The Journal of clinical investigation, 121(5), 2025-2036.
Puigserver, P., Rhee, J., Lin, J., Wu, Z., Yoon, J. C., Zhang, C.-Y., . . . Spiegelman, B. M. (2001). Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Molecular cell, 8(5), 971-982.
Ramana, C., Gil, M., Schreiber, R., & Stark, G. (2002). Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends in Immunology, 23(2), 96-101.
Ramprasad, M. P., Terpstra, V., Kondratenko, N., Quehenberger, O., & Steinberg, D. (1996). Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proceedings of the National Academy of Sciences, 93(25), 14833-14838.
Ricciarelli, R., Zingg, J.-M., & Azzi, A. (2000). Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation, 102(1), 82-87.
Rios, F. J., Gidlund, M., & Jancar, S. (2011). Pivotal role for platelet-activating factor receptor in CD36 expression and oxLDL uptake by human monocytes/macrophages. Cellular Physiology and Biochemistry, 27(3-4), 363-372.
Roger, V. L., Go, A. S., Lloyd-Jones, D. M., Adams, R. J., Berry, J. D., Brown, T. M., . . . Stroke Stat, S. (2011). Executive summary: heart disease and stroke statistics-2011 update A report from the American Heart Association. Circulation, 123(4), 459-463. doi: 10.1161/CIR.0b013e31820c7a50
Roskoski, R. (2012). ERK1/2 MAP kinases: structure, function, and regulation. Pharmacological research, 66(2), 105-143.
Ross, R. (1986). The pathogenesis of atherosclerosis—an update. New England Journal of Medicine, 314(8), 488-500.
Ross, R. (1995). Cell biology of atherosclerosis. Annual review of physiology, 57(1), 791-804.
Santulli, G. (2013). Epidemiology of cardiovascular disease in the 21st century: updated numbers and updated facts. J Cardiovasc Dis, 1(1), 1-2.
Schepetkina, A. (2013). Mechanisms of 7,8-dihydroneopterin protection of macrophages from cytotoxicity. University of Canterbury.
Schild, R., Sonnenberg-Hirche, C., Schaiff, W., Bildirici, I., Nelson, D., & Sadovsky, Y. (2006). The kinase p38 regulates peroxisome proliferator activated receptor-γ in human trophoblasts. Placenta, 27(2), 191-199.
Shao, D., Rangwala, S. M., Bailey, S. T., Krakow, S. L., Reginato, M. J., & Lazar, M. A. (1998). Interdomain communication regulating ligand binding by PPAR-γ. Nature, 396(6709), 377-380.
Shchepetkina, A. (2013). Mechanisms of 7,8-dihydroneopterin protection of macrophages from cytotoxicity. University of Canterbury.
Shiffman, D., Mikita, T., Tai, J. T., Wade, D. P., Porter, J. G., Seilhamer, J. J., . . . Lawn, R. M. (2000). Large scale gene expression analysis of cholesterol-loaded macrophages. Journal of Biological Chemistry, 275(48), 37324-37332.
Stary, H. C., Chandler, A. B., Dinsmore, R. E., Fuster, V., Glagov, S., Insull, W., . . . Wissler, R. W. (1995). A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation, 92(5), 1355-1374.
Steinberg, D. (1989). Beyond cholesterol modification of low density lipoprotein that increase its atherogenecity. New eng J, 320, 915-924.
Steinberg, D. (2009). The LDL modification hypothesis of atherogenesis: an update. Journal of lipid research, 50(Supplement), S376-S381.
Steinbrecher, P. (1991). Role of lipoprotein peroxidation in the pathogenesis of atherosclerosis. Clinical cardiology, 14(11), 865-867.
Stocker, R., & Keaney, J. F. (2004). Role of oxidative modifications in atherosclerosis. Physiological reviews, 84(4), 1381-1478.
Struzik, J., Szulc-Dabrowska, L., & Niemialtowski, M. (2014). Modulation of NF-кB transcription factor activation by Molluscum contagiosum virus proteins. Postepy Hig Med Dosw (online), 68, 129-136.
Swerlick, R. A., Lee, K., Wick, T., & Lawley, T. (1992). Human dermal microvascular endothelial but not human umbilical vein endothelial cells express CD36 in vivo and in vitro. The Journal of Immunology, 148(1), 78-83.
Talle, M., Rao, P., Westberg, E., Allegar, N., Makowski, M., Mittler, R., & Goldstein, G. (1983). Patterns of antigenic expression on human monocytes as defined by monoclonal antibodies. Cellular immunology, 78(1), 83-99.
Tian, G.-P., & Tang, C.-K. (2014). NF-κB suppresses the expression of ATP-binding cassette transporter A1/G1 by regulating SREBP-2 and miR-33a in mice. International journal of cardiology, 171, e93-e95.
Todd, J. L., Tanner, K. G., & Denu, J. M. (1999). Extracellular regulated kinases (ERK) 1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase via a novel role in down-regulating the ERK pathway. Journal of Biological Chemistry, 274(19), 13271-13280.
Tontonoz, P., Hu, E., & Spiegelman, B. M. (1994). Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell, 79(7), 1147-1156.
Tontonoz, P., Nagy, L., Alvarez, J. G., Thomazy, V. A., & Evans, R. M. (1998). PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell, 93(2), 241-252.
Tontonoz, P., & Spiegelman, B. M. (2008). Fat and beyond: the diverse biology of PPARγ. Annu. Rev. Biochem., 77, 289-312.
Viana, M., Villacorta, L., Bonet, B., Indart, A., Munteanu, A., Sanchez-Vera, I., . . . Zingg, J. (2005). Effects of aldehydes on CD36 expression. Free radical research, 39(9), 973-977.
Wachter, H., Fuchs, D., Hausen, A., Reibnegger, G., Weiss, G., Werner, E., & Werner-Felmayer, G. (1992). Neopterin: biochemistry-methods-clinical application: Walter de Gruyter.
Wang, N., Tabas, I., Winchester, R., Ravalli, S., Rabbani, L. E., & Tall, A. (1996). Interleukin 8 is induced by cholesterol loading of macrophages and expressed by macrophage foam cells in human atheroma. Journal of Biological Chemistry, 271(15), 8837-8842.
Weiss, G., Fuchs, D., Hausen, A., Reibnegger, G., Werner, E. R., Werner-Felmayer, G., . . . Wachter, H. (1993). Neopterin modulates toxicity mediated by reactive oxygen and chloride species. FEBS letters, 321(1), 89-92.
Werner-Felmayer, G., Baier-Bitterlich, G., Fuchs, D., Hausen, A., Murr, C., Reibnegger, G., . . . Wachter, H. (1995). Detection of bacterial pyrogens on the basis of their effects on gamma interferon-mediated formation of neopterin or nitrite in cultured monocyte cell lines. Clinical and diagnostic laboratory immunology, 2(3), 307-313.
Werner-Felmayer, G., Werner, E. R., Fuchs, D., Hausen, A., Reibnegger, G., & Wachter, H. (1990). Neopterin formation and tryptophan degradation by a human myelomonocytic cell line (THP-1) upon cytokine treatment. Cancer research, 50(10), 2863-2867.
Werner, E. R., Werner-Felmayer, G., Fuchs, D., Hausen, A., Reibnegger, G., Yim, J., . . . Wachter, H. (1990). Tetrahydrobiopterin biosynthetic activities in human macrophages, fibroblasts, THP-1, and T 24 cells. GTP-cyclohydrolase I is stimulated by interferon-gamma, and 6-pyruvoyl tetrahydropterin synthase and sepiapterin reductase are constitutively present. Journal of Biological Chemistry, 265(6), 3189-3192.
Williams, K. J., & Tabas, I. (1995). The response-to-retention hypothesis of early atherogenesis. Arteriosclerosis, thrombosis, and vascular biology, 15(5), 551-561.
Wu, H.-M., Wang, J., Zhang, B., Fang, L., Xu, K., & Liu, R.-Y. (2016). CpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus-stimulated macrophage. Life sciences, 161, 51-59.
Xue, J.-h., Yuan, Z., Wu, Y., Liu, Y., Zhao, Y., Zhang, W.-p., . . . Kishimoto, C. (2010). High glucose promotes intracellular lipid accumulation in vascular smooth muscle cells by impairing cholesterol influx and efflux balance. Cardiovascular research, 86(1), 141-150.
Yang, X., Yao, H., Chen, Y., Sun, L., Li, Y., Ma, X., . . . Han, J. (2015). Inhibition of glutathione production induces macrophage CD36 expression and enhances cellular-oxidized low density lipoprotein (oxLDL) uptake. Journal of Biological Chemistry, 290(36), 21788-21799.
Yang, Y.-t. T., Whiteman, M., & Gieseg, S. (2012). Intracellular glutathione protects human monocyte-derived macrophages from hypochlorite damage. Life sciences, 90(17), 682-688.
Ye, X., Jiang, X., Guo, W., Clark, K., & Gao, Z. (2013). Overexpression of NF-κB p65 in macrophages ameliorates atherosclerosis in apoE-knockout mice. American Journal of Physiology-Endocrinology and Metabolism, 305(11), E1375-E1383.
Yoshida, H., Kondratenko, N., Green, S., Steinberg, D., & Quehenberger, O. (1998). Identification of the lectin-like receptor for oxidized low-density lipoprotein in human macrophages and its potential role as a scavenger receptor. Biochemical Journal, 334(1), 9-13.
Yoshizumi, M., Abe, J., Haendeler, J., Huang, Q., & Berk, B. C. (2000). Src and Cas mediate JNK activation but not ERK1/2 and p38 kinases by reactive oxygen species. Journal of Biological Chemistry, 275(16), 11706-11712.
Yoshizumi, M., Kim, S., Kagami, S., Hamaguchi, A., Tsuchiya, K., Houchi, H., . . . Tamaki, T. (1998). Effect of endothelin‐1 (1‐31) on extracellular signal‐regulated kinase and proliferation of human coronary artery smooth muscle cells. British journal of pharmacology, 125(5), 1019-1027.
Yoshizumi, M., Tsuchiya, K., & Tamaki, T. (2001). Signal transduction of reactive oxygen species and mitogen-activated protein kinases in cardiovascular. The Journal of Medical Investigation, 48, 11-24.
Yu, X.-H., Jiang, H.-L., Chen, W.-J., Yin, K., Zhao, G.-J., Mo, Z.-C., . . . Zhang, D.-W. (2011). Interleukin-18 and interleukin-12 together downregulate ATP-binding cassette transporter A1 expression through the interleukin-18R/nuclear factor-κB signaling pathway in THP-1 macrophage-derived foam cells. Circulation journal: official journal of the Japanese Circulation Society, 76(7), 1780-1791.
Yun, M. R., Im, D. S., Lee, S. J., Woo, J. W., Hong, K. W., Bae, S. S., & Kim, C. D. (2008). 4-hydroxynonenal contributes to macrophage foam cell formation through increased expression of class A scavenger receptor at the level of translation. Free Radical Biology and Medicine, 45(2), 177-183.
Yvan-Charvet, L., Wang, N., & Tall, A. R. (2010). Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arteriosclerosis, thrombosis, and vascular biology, 30(2), 139-143.
Zamora, C., Cantó, E., Nieto, J. C., Ortiz, M. A., Juarez, C., & Vidal, S. (2012). Functional consequences of CD36 downregulation by TLR signals. Cytokine, 60(1), 257-265.
Zeng, Y., Tao, N., Chung, K.-N., Heuser, J. E., & Lublin, D. M. (2003). Endocytosis of oxidized low density lipoprotein through scavenger receptor CD36 utilizes a lipid raft pathway that does not require caveolin-1. Journal of Biological Chemistry, 278(46), 45931-45936.
Zhao, G.-j., Mo, Z.-C., Tang, S.-L., Ouyang, X.-P., He, P.-p., Lv, Y.-c., . . . Shi, J.-F. (2014). Chlamydia pneumoniae negatively regulates ABCA1 expression via TLR2-Nuclear factor-kappa B and miR-33 pathways in THP-1 macrophage-derived foam cells. Atherosclerosis, 235(2), 519-525.
Zhao, J. F., Ching, L. C., Huang, Y. C., Chen, C. Y., Chiang, A. N., Kou, Y. R., . . . Lee, T. S. (2012). Molecular mechanism of curcumin on the suppression of cholesterol accumulation in macrophage foam cells and atherosclerosis. Molecular nutrition & food research, 56(5), 691-701.
Zhao, M., Liu, Y., Wang, X., New, L., Han, J., & Brunk, U. T. (2002). Activation of the p38 MAP kinase pathway is required for foam cell formation from macrophages exposed to oxidized LDL. Apmis, 110(6), 458-468.
Zhu, Y., Alvares, K., Huang, Q., Rao, M. S., & Reddy, J. (1993). Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. Journal of Biological Chemistry, 268(36), 26817-26820.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biology"
Biology is the scientific study of the natural processes of living organisms or life in all its forms. including origin, growth, reproduction, structure, and behaviour and encompasses numerous fields such as botany, zoology, mycology, and microbiology.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: