Role of Prorenin in Placentation
Info: 7596 words (30 pages) Dissertation
Published: 9th Mar 2021
Tagged: Biomedical Science
Novel insights into the molecular pathways that underpin trophoblast proliferation, invasion and migration and placental angiogenesis.
Keywords: Trophoblast, placenta, renin angiotensin system, angiogenesis, cell invasion, pregnancy
__________________________________________________________________________________
ABSTRACT
Preeclampsia is a significant and frequently occurring hypertensive disorder that complicates human pregnancy. It is a multisystem disorder that is one of the main causes of maternal and perinatal morbidity and mortality globally. While the aetiology, pathogenesis and pathophysiology of preeclampsia are not precisely known, one of the major causes of preeclampsia is impaired placentation. Successful development of the placenta requires a complex interplay between proliferation of trophoblast cells and their invasion of uterine tissues and remodelling of uterine spiral arterioles. There is strong evidence to suggest that angiotensin II, a product of the renin-angiotensin system, stimulates trophoblast proliferation, invasion and placental angiogenesis when it acts through its type 1 receptor. However there is little known regarding the upstream molecules involved in the renin angiotensin system, such as the precursor to active renin, prorenin. The aim of this review is to investigate the function of the renin-angiotensin system in placentation, and in particular to elucidate the role of prorenin and the interaction with its receptor that underpin trophoblast proliferation, invasion and migration in the developing placenta.
__________________________________________________________________________________
1.1 Introduction
The placenta is a highly specialised organ of pregnancy that supports normal growth and development of the fetus (1). Growth and function of the placenta are tightly regulated and coordinated to ensure the exchange of nutrients, gases and waste products between the maternal and fetal circulatory systems occurs effectively (1). Impaired placentation can lead to the development of intrauterine growth restriction (IUGR) and preeclampsia (2,3). The systemic renin angiotensin system (RAS) is an endocrine system that acts to regulate blood pressure, tissue perfusion and fluid balance (4). However a local tissue-based RAS also exists within the placenta, which functions separately from the circulating system (5). Angiotensin II (Ang II) is the major hormone product that results from the RAS, and levels of this hormone have been found to be highest in early gestation (5), where it acts via the proliferative angiotensin II type 1 receptor (AT1R), to stimulate vasoconstriction, cell proliferation and angiogenesis (6,7). Further to this, is the role of prorenin, the precursor to active renin, which until recently was considered to have little biological activity (8). Prorenin can directly stimulate intracellular signalling via the prorenin receptor ((P)RR) or can be activated by the receptor to produce Angiotensin I (Ang I) from angiotensinogen. Through the RAS, Ang II can be generated from Ang I, and through prorenin/(P)RR, the functional effects of a systemic RAS can be implicated at a tissue level within the placenta. It is known that the developing placenta contains high levels of prorenin and (P)RR and it is proposed that the actions of this pathway – which include tissue growth, migration, invasion and angiogenesis – are critical for placentation (9).
2.1 The Placenta
2.1.1 PLACENTAL DEVELOPMENT
Development of the placenta is a highly regulated process that is fundamental for the maintenance of a healthy pregnancy and normal fetal growth (10). The development of the placenta and fetus begins at the time of fertilisation. After approximately 5 days of growth, the morula becomes a blastocyst as fluid accumulates and polarisation of the cells occur (10). The blastocyst consists of an outer layer of cells referred to as the trophectoderm, which will eventually form the placenta and fetal membranes. The blastocyst also contains an inner cell mass that will form the embryo, and a fluid filled cavity that swells to hatch the blastocyst out of the zona pellucida shell (10,11). Upon implantation of the blastocyst into the uterine lining, the trophoblast cells of the trophectoderm proliferate and differentiate into 2 cell layers – the outer syncytiotrophoblast and the inner cytotrophoblast cells (12). The syncytiotrophoblast layer is formed from fusion of the underlying cytotrophoblasts to become a single multinucleated syncytiotrophoblast cell. The syncytiotrophoblast covers the entire surface of the placenta and is in direct contact with maternal blood (13). This cellular layer is central to coordinating biomolecular interactions between the fetus and the mother (13). Beneath the syncytiotrophoblast is a layer of cytotrophoblast cells, which are considered the stem cells of the placenta (10,13). These progenitor cells proliferate throughout gestation, differentiating along two pathways to form either villous cytotrophoblasts or extravillous cytotrophoblasts (10,14). In the villous pathway, cytotrophoblast cells fuse to form the multinucleated syncytiotrophoblast. In the extravillous pathway, cytotrophoblast cells acquire an invasive phenotype and differentiate into either (1) interstitial extravillous trophoblasts, which invade the decidua and a portion of the myometrium, or (2) endovascular extravillous trophoblasts, which remodel the maternal vasculature (14). These differentiations are influenced by a combination of factors including oxygen tension, hormones, growth factors, microRNAs and other signalling factors (14).
2.1.1.1 Formation of the chorionic villi
The definitive placenta consists of a fetal portion, derived from the chorion, and a maternal portion, formed by the decidua. In order to gain access to the maternal blood, the embryonic trophoblasts must invade the decidualised endometrial stroma of the uterine wall (15). At approximately 9 days post fertilisation, fluid-filled spaces called lacunae develop within the syncytiotrophoblast and fill with blood from ruptured maternal capillaries and endometrial glandular secretions (16). This association with the maternal vasculature is the first step towards the establishment of the uteroplacental circulation. Following this, fingers of rapidly dividing cytotrophoblast cells project into the syncytiotrophoblast, forming “primary” villi (15). 14 days after fertilisation an extraembryonic space, the chorionic cavity, is established. This cavity is bound by extra-embryonic somatic mesoderm underlying the cytotrophoblast cells. When the primary villi become filled with mesenchyme from this mesoderm, they are termed secondary villi (15). The mesenchymal core differentiates to form fetal capillaries and when this process is completed, the embryonic blood vessels enter the villi to form the feto-placental circulation, they are then termed tertiary chorionic villi (15,16). By approximately day 21 post fertilisation, the primitive embryonic heart begins to pump blood through the capillaries of the chorionic villi (16). In its final form the placenta consists of stem villi that extend from the chorionic plate. These villi either float within the intervillous space (maternal blood space) as terminal villi or extend and attach to the maternal decidua as anchoring villi (15,16) (Fig. 1).
2.1.1.2 Invasion of spiral arteries
An important final step in establishing the uteroplacental circulation involves the invasion of the spiral arteries in the uterine wall by extravillous trophoblasts. At approximately 4 weeks of gestation, extravillous trophoblasts, which have broken through the overlying syncytium, expand in columns with proliferative trophoblasts at the base and invasive trophoblasts at the distal end (10) (Fig. 1). The invasive extravillous trophoblasts that penetrate the decidua are termed interstitial extravillous trophoblasts, while the extravillous trophoblasts, which invade and remodel the spiral arteries are termed endovascular extravillous trophoblasts (10) (Fig. 1). Endovascular penetration by trophoblasts involves replacement of the endothelium and much of the tunica media as the narrow spiral arteries transform into wide uteroplacental arteries (10,15) (Fig. 2). These dilated vessels anastomose with endometrial veins to form maternal sinusoids, which act to deliver blood into the low resistance lacunar network system (10). If the cytotrophoblasts do not completely invade the decidua or if they fail to convert the spiral arteries to low resistance vessels, maternal blood flow to the placenta will be insufficient. This occurrence promotes the secretion of vasoactive substances that can result in serious maternal complications of pregnancy conditions such as gestational hypertension and preeclampsia (15).
2.1.2 PLACENTAL FUNCTION
The placenta is a short-lived maternal-fetal organ essential for the normal development of the fetus (17). The interaction between the chorionic villi and the maternal blood spaces allows for metabolism and transport of substances between maternal and fetal circulatory systems (1,18). During pregnancy, the placenta has nutritive, immune, endocrine and excretory functions (17).
During the majority of the first trimester nutrition is histotrophic, whereby trophoblasts phagocytose endometrial glandular secretions in order to produce glycogen (1). At approximately 10-12 weeks gestation, maternal blood is in contact with the terminal villi of the placenta and this allows for the transport of respiratory gases, nutrients and waste products across the placental membrane (1). The high permeability of this membrane for gases allows for rapid passive diffusion of oxygen from maternal to fetal blood, and of carbon dioxide from fetal to maternal blood (1,19,20). Nutrients such as glucose, amino acids, fatty acids and electrolytes are required to support fetal growth (18). These substances are obtained from the maternal blood and their transport across the placenta is most commonly via facilitated diffusion (1). Waste products such as urea, uric acid and creatinine are excreted into the maternal blood stream by simple diffusion.
The placenta also functions as an immunological barrier between the mother and fetus. Numerous strategies are employed by trophoblast cells to protect the embryo and the extraembryonic membranes. Altered human leukocyte antigen (HLA) expression, synthesis of immunosuppressive molecules and expression of high levels of complementary regulatory proteins are all methods that these trophoblast cells use to avoid maternal immune cells and antibody-mediated cell destruction (21).
During pregnancy, the placenta also plays a role as an important endocrine organ. It secretes both peptide and steroid hormones, which act to maintain pregnancy and prepare for parturition and lactation (18). Human chorionic gonadotrophin (hCG) and human placental lactogen (hPL) are both synthesised by the syncytiotrophoblast, and are two of the main peptide hormones secreted by the placenta (1,18,20). hPL is a growth-like hormone that acts to promote breast development for lactation and it also plays a role in prioritising maternal blood glucose towards the fetus (18,20). During early pregnancy hCG acts to prolong the life of the corpus luteum in order for it to maintain progesterone secretion. After 6-8 weeks of gestation the placenta carries the role of progesterone production and the secretion of hCG decreases (18,22). The steroid hormone progesterone that is secreted by the placenta functions to maintain the endometrium, reduce myometrial activity and suppress maternal immunological responses to fetal antigens (18). In addition to progesterone, the placenta produces increasing amounts of estriol in order to stimulate uterine growth and development of mammary glands (20).
2.1.3 PREGNANCY COMPLICATIONS ARISING DUE TO POOR PLACENTATATION
Due to the placenta’s role in immune, endocrine, respiratory, nutritive and excretory functions (23), correct development and function of the placenta are vital for embryonic and fetal growth (24). Poor placentation has been implicated in a variety of pregnancy complications including miscarriage, preterm birth, intrauterine growth restriction and preeclampsia.
Preeclampsia is a multisystem disorder that complicates 3-8% of pregnancies in Western countries and represents a major cause of morbidity and mortality worldwide (25). The condition is unique to human pregnancy and is characterised by hypertension and proteinuria (13,14). In the mother, preeclampsia can be life threatening, however it may also cause the development of conditions such as chronic hypertension, ischemic heart disease and stroke later in life (25,26). Neonatal complications of preeclampsia include perinatal death, preterm delivery and fetal growth restriction. In adult life, children born after preeclamptic pregnancies are at an increased risk of stroke, coronary heart disease and metabolic syndrome (25,27).
The limited invasion of trophoblast cells into the uterus and subsequent failure of the remodelling of maternal spiral arteries is believed to be a primary cause of preeclampsia (14) (Fig. 2). Brosen et al. investigated the placentas of women with preeclampsia and discovered that cytotrophoblast invasion of the uterus is superficial, and the endovascular invasion does not proceed beyond the terminal portions of the spiral arterioles. They therefore described preeclampsia as being associated with “shallow” cytotrophoblast invasion (13,28). De Wolf et al. observed that extravillous cytotrophoblast invasion of the uterine spiral arterioles is often incomplete by 22 weeks gestation and that spiral arteries fail to lose their muscular elastic components, therefore inhibiting their conversion to “low resistance” capacitance vessels (13,29). Figure 2 illustrates abnormal spiral artery remodelling that results in low flow and high resistance in cases of preeclamptic placentae. Oxidative stress, release of vasoactive substances into the maternal circulation, as well as irregular expression of signalling molecules have been proposed to be involved in the pathogenesis of preeclampsia (14). However, despite extensive investigations into the pathogenesis of preeclampsia it should be emphasised that the causes of this multifactorial disorder remain unknown. While poor placentation has been theorised to be a key cause of preeclampsia, it is now regarded as a strong predisposing factor to the illness (27). And it is the mechanisms that underpin this poor placentation that are the focus for future investigations into the pathogenesis of pregnancy complications such as preeclampsia.
3.1 Renin Angiotensin System (RAS)
3.1.1 RAS OVERVIEW
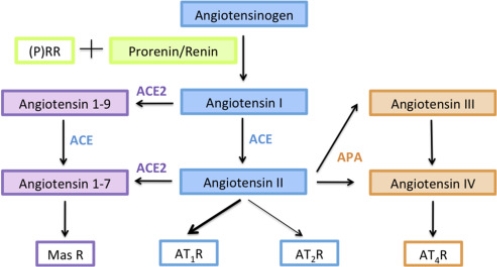
Since the discovery of the renin-angiotensin system (RAS) in 1898 with studies made by Tigerstedt and Bergman, this system has been extensively investigated (30,31). The RAS is a hormonal cascade that functions in the homeostatic control of blood pressure, tissue perfusion and fluid balance (4,6). The classical view of the RAS is that renin, synthesised in the kidney, acts in the circulation on a substrate, angiotensinogen, synthesised in the liver, to yield angiotensin I (Ang I) (32). Angiotensin converting enzyme (ACE), present in most tissues, cleaves Ang I to generate Angiotensin II (Ang II) (6). Ang II then acts via one of two angiotensin receptors (AT1R or AT2R) (Fig. 3). Ang II acting via the AT1R mediates vasoconstriction, thirst, cellular growth, migration and release of vasopressin and aldosterone (7). This process stimulates angiogenesis, cell proliferation and invasion (6). On the other hand, the role of the AT2R receptor in physiology is only just being uncovered (33). Studies have highlighted that the AT2R plays an important functional role in prenatal development, and in the adult AT2R-mediated actions have been shown to counteract the effects of AT1R such as cell proliferation in vitro (34) and in vivo (35,36).
More recently, an expanded view of the RAS has gradually emerged. It is now evident that in addition to the classical circulating RAS outlined above, there is also a local RAS in most organs and tissues (7). The tissue RASs, e.g. in the kidney, heart, brain and reproductive organs, have local effects involving proliferation, growth, protein synthesis and other organ-specific functions (7,37). Therefore the view of RAS as an endocrine system can be expanded to include its role in paracrine and in intracrine systems (7).
Until recently, prorenin, the precursor of active renin was thought to have limited or no biological activity (8). However the discovery that the (pro)renin receptor ((P)RR) can bind both prorenin and renin to generate Ang I from angiotensinogen has expanded the view of prorenins role in the local RAS (Fig. 3). Binding of prorenin to the (P)RR leads to non-proteolytic activation of prorenin, ultimately increasing contractility, hypertrophy and fibrosis (7). Additionally, binding of prorenin or renin to the (P)RR can activate intracellular signalling pathways independent of Ang II formation (8). This is highlighted in studies conducted by Feldt et al, where Ang II synthesis was blocked by aliskiren, and the ability of prorenin to bind and signal through the (P)RR was evaluated. Aliskiren had no (P)RR blocking effect, and did not inhibit the kinase activity of prorenin and the (P)RR (38). Therefore, if (P)RR is present, prorenin could activate the placental RAS, as well as many other tissue RAS’s, through the production of Ang II and also through Ang II independent pathways (8) (Fig. 3).
3.1.2 PLACENTAL RAS
It has been speculated by many authors that the placental RAS may influence placental function, since different components of the RAS are present in the conceptus from 6 weeks of gestation until term (39-42). More specifically, the human placenta expresses all components of the RAS that lead to the generation of Ang II (6). The various RAS-proteins are expressed differently in the different areas of this organ. Angiotensinogen, renin, Ang I, Ang II, ACE, AT1R and AT2R have been localised to the maternal decidua (40,42,43). Angiotensin and renin were additionally found in fetal capillaries (40,42). The AT1R, which is highly expressed in the placenta, has been detected in cytotrophoblast and syncytiotrophoblast cells as well as in fetal capillaries. On the other hand the AT2R is expressed at much lower levels and is predominantly located in fetal tissues (41,42). Ang II binding to AT2R increases apoptosis, causes vasodilation and is involved in fetal tissue development (44).
Studies made by Pringle et al demonstrate that the expression of the placental RAS changes across gestation (6). Renin mRNA and angiotensinogen mRNA levels were found to be highest in early gestation, while ACE expression is highest in term placentae (6). Studies investigating the expression of the AT1R and AT2R have reflected conflicting results. Pringle et al showed evidence that levels of AT1R mRNA were highest in early gestation samples compared to late and term samples. Contrastingly studies by Petit et al highlight that both AT1R mRNA and protein levels increase from the first trimester onwards, reaching the highest levels at term (45). Ultimately, as suggested by Kalenga et al, perhaps the concentration of Ang II regulates AT1R expression. This is suggested as a positive correlation between Ang II concentration and the number of AT1Rs in the human placenta has been observed (43).
In early pregnancy prorenin has been located in the syncytiotrophoblast as well as the villous and extravillous cytotrophoblasts of the human placenta (6). At term, prorenin, renin and the (P)RR are predominately found in syncytiotrophoblast (6). Thus far there have been no studies to determine if protein levels of prorenin, active renin or (P)RR in the placenta change throughout gestation. However since renin mRNA is highest in the first trimester it is suggested that placental prorenin production also peaks early in pregnancy (6). The co-localisation of (P)RR and prorenin in the placenta combined with their high mRNA expression in early gestation, when placental invasion is greatest, signifies that prorenin could act via the placental RAS and regulate trophoblast migration and spiral artery remodelling.
3.1.3 FUNCTIONS OF THE RAS IN PLACENTAL DEVELOPMENT
It is known that trophoblasts are rich in AT1R and thus are responsive to changes in Ang II concentrations that occur during pregnancy (46). Pan et al. utilised an in vitro cell model to highlight the involvement of local RAS components in physiologic and pathophysiological processes during pregnancy (47). Pan et al emphasised that Ang II is involved in the regulation of uteroplacental vascular resistance and blood flow. Low Ang II concentrations increase uteroplacental blood flow, whereas high concentrations of Ang II reduce blood flow. Additionally, Ang II acting through the AT1R also stimulates plasminogen activator inhibitor-1 (PAI-1) gene expression, and thus inhibits human trophoblast invasion, a process involved in the physiological union between maternal and fetal circulatory systems (47). Cell proliferation was also stimulated through the Ang II/AT1R interaction (8). In light of studies such as those conducted by Xia et al (48) and George et al (49), it is highly likely that the placental RAS is a key mediator of placental angiogenesis, trophoblast proliferation and invasion (8). It is also thought that deregulation of expression of components of the placental RAS could influence placental development and lead to placental insufficiency (8).
However, limited research exists on the functional role of prorenin in placentation. Pioneer studies involving the (P)RR portray the ability of this newly discovered component of the RAS to promote Ang II formation via binding to renin or prorenin (50). Rodrigues et al explored the functional role of (P)RR in placentation, and discovered that by knocking down the receptor in an extravillous trophoblast cell line, cell proliferation and migration rate’s dropped significantly (9). It is now proposed that the (P)RR is vital for placentation, and that it’s effects can be mediated by both RAS dependent and independent mechanisms. This (P)RR-induced prorenin activation could explain how prorenin exerts pathological effects in placentation, and deregulation of this system could result in impaired placentation (38).
3.1.4 PLACENTAL RAS IN PREECLAMPSIA
Pregnancy is characterised by an increase in many of the different components of the circulating renin-angiotensin system (RAS). However, the physiological mechanisms of stimulated RAS activity during pregnancy are still under investigation. Even less understood is how this system may be altered in hypertensive disorders of pregnancy such as preeclampsia (51). In women with preeclampsia, dysregulation of both the circulating and placental RAS has been observed when compared to women with uncomplicated pregnancies. Lower plasma levels of renin, aldosterone, Ang I and Ang II have been reported in patients suffering from preeclampsia (52).
However whether this is a cause or a consequence of the disease is not clearly known (8). Raised levels of renin and prorenin in placental tissues of women with preeclampsia is suggestive that both prorenin and renin have cellular effects independent of Ang II generation (53). Therefore, it is suggested that the physiological action of the tissue RAS, rather than the circulating RAS, may play a more important role in the aetiology and pathogenesis of preeclampsia (54). The possible biological effect(s) of the extracellular prorenin are unknown, however since angiotensin-peptides have a number of trophic effects on both vascular and non-vascular tissues, regulation of uteroplacental prorenin by autocrine, paracrine or endocrince signally may be critical to normal fetal and placental development (55).
4.1 Conclusion
Despite extensive research, the pathological cause of preeclampsia remains unknown. The studies described in this review provide evidence that both the circulating and uteroplacental RAS play an important role in understanding the mechanisms responsible for the development of the placenta. Due to its high expression during early placentation, and the expression of key molecules of the prorenin/(P)RR and Ang II/AT1R pathway in invading cells, it suggests that the placental RAS plays a pivotal role in placental angiogenesis, proliferation and trophoblast migration. Furthermore, placental prorenin, activated by binding to the prorenin receptor or through proteolytic activation by endogenous proteases may react with maternal and fetal circulating angiotensinogen. However, in order to gain a comprehensive understanding of the mechanisms responsible for regulation of RAS components in normal pregnancy and preeclampsia, further investigation is needed.
5.1 Research project
Our research project aims to provide insights into the molecular pathways that underpin trophoblast proliferation, invasion and placental angiogenesis. We aim to elucidate the functional role of prorenin, its interactions with the prorenin receptor and how this drives placentation. In order to achieve this, a first trimester trophoblast cell line (HTR-8/SVneo) will be transfected with prorenin (REN) siRNA to knockdown endogenous prorenin expression. The transfection efficiency will be assessed by qPCR and western blotting. Proliferation, invasion and migration will be assessed using the xCELLigence real time cell analysis system. This project intends to show that prorenin is essential for proliferation, migration and invasion in development of the placenta.
6.1 Acknowledgements
I would like to acknowledge Dr Kirsty Pringle and Samantha Rodrigues of the Mothers and Babies Research Centre at the Hunter Medical Research Institute, for their assistance in the editing of this literature review.
7.1 Conflicts of interest
The authors declare that they have no conflicts of interests with the contents of this article.
8.1 References
1. Gude, N. M., Roberts, C. T., Kalionis, B., and King, R. G. (2004) Growth and function of the normal human placenta. Thrombosis Research 114, 397-407
2. Shah, D. M. (2005) Role of the renin-angiotensin system in the pathogenesis of preeclampsia. American journal of physiology. Renal physiology 288, F614-625
3. Scifres, C. M., and Nelson, D. M. (2009) Intrauterine growth restriction, human placental development and trophoblast cell death. The Journal of Physiology 587, 3453-3458
4. Atlas, S. A. (2007) The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. Journal of managed care pharmacy : JMCP 13, 9-20
5. Herse, F., Dechend, R., Harsem, N. K., Wallukat, G., Janke, J., Qadri, F., Hering, L., Muller, D. N., Luft, F. C., and Staff, A. C. (2007) Dysregulation of the Circulating and Tissue-Based Renin-Angiotensin System in Preeclampsia. Hypertension (Dallas, Tex. : 1979) 49, 604
6. Pringle, K. G., and Lumbers, E. R. (2013) The placental renin angiotensin system. Nova Science Publishers
7. Fyhrquist, F., and Saijonmaa, O. (2008) Renin-angiotensin system revisited. Journal of Internal Medicine 264, 224-236
8. Pringle, K. G., Tadros, M. A., Callister, R. J., and Lumbers, E. R. (2011) The expression and localization of the human placental prorenin/renin-angiotensin system throughout pregnancy: Roles in trophoblast invasion and angiogenesis? Placenta 32, 956-962
9. Rodrigues, S., Morosin, S., Delforce, S., Mohammed, R., Lumbers, E., and Pringle, K. (2017) The placental prorenin/prorenin receptor system. Placenta 57, 298
10. Roberts, V., and Myatt, L. (2017) Placental development and physiology 19-05-2017 Ed., UpToDate
11. Redman, C. W. (1986) Immunology of the placenta. Clinics in obstetrics and gynaecology 13, 469-499
12. Burrows, T. D., King, A., and Loke, Y. W. (1996) Trophoblast migration during human placental implantation. Hum Reprod Update 2, 307-321
13. Wang, Y., and Zhao, S. (2010) Integrated Systems Physiology: from Molecules to Function to Disease. in Vascular Biology of the Placenta, Morgan & Claypool Life Sciences
Copyright (c) 2010 by Morgan & Claypool Life Sciences., San Rafael (CA). pp
14. Ji, L., Brkić, J., Liu, M., Fu, G., Peng, C., and Wang, Y.-L. (2013) Placental trophoblast cell differentiation: Physiological regulation and pathological relevance to preeclampsia. Molecular Aspects of Medicine 34, 981-1023
15. Witkin, J. W. (2004) Formation and Role of Placenta Dept. Anatomy & Cell Biology, P&S 12-432
16. Stirrat, G. M. (1989) The Immunology of Diseases of Pregnancy. in Immunology of Pregnancy and its Disorders (Stern, C. M. M. ed.), Springer Netherlands, Dordrecht. pp 143-164
17. Verma, U., and Verma, N. (2013) An overview of development, function, and diseases of the placenta. in The Placenta: Development, Function and Diseases. pp 1-30
18. Donnelly, L., and Campling, G. (2008) Functions of the placenta. Anaesthesia & Intensive Care Medicine 9, 124-127
19. Murphy, V. E., Smith, R., Giles, W. B., and Clifton, V. L. (2006) Endocrine Regulation of Human Fetal Growth: The Role of the Mother, Placenta, and Fetus. Endocrine Reviews 27, 141-169
20. Sadler, T. W., and Langman, J. (2012) Langman’s medical embryology, Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia
21. Abrahams, V. M. (2017) Immunology of the maternal-fetal interface 31-01-2017 Ed., UpToDate
22. Evain-Brion, D., and Malassine, A. (2003) Human placenta as an endocrine organ. Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research Society 13 Suppl A, S34-37
23. Donnelly, L., and Campling, G. (2008) Functions of the placenta. Anaesthesia & intensive care medicine. 9, 124-127
24. Hill, M. (2017) Embryology Placenta – Abnormalities UNSW Embryology UNSW Embryology pp
25. Uzan, J., Carbonnel, M., Piconne, O., Asmar, R., and Ayoubi, J.-M. (2011) Pre-eclampsia: pathophysiology, diagnosis, and management. Vascular Health and Risk Management 7, 467-474
26. Ramsay, J. E., Stewart, F., Greer, I. A., and Sattar, N. (2003) Microvascular dysfunction: a link between pre-eclampsia and maternal coronary heart disease. BJOG: An International Journal of Obstetrics and Gynaecology 110, 1029-1031
27. Sibai, B., Dekker, G., and Kupferminc, M. (2005) Pre-eclampsia. The Lancet 365, 785-799
28. Brosens, I. A., Robertson, W. B., and Dixon, H. G. (1970) The role of the spiral arteries in the pathogenesis of pre-eclampsia. The Journal of pathology 101, Pvi
29. De Wolf, F., Robertson, W. B., and Brosens, I. (1975) The ultrastructure of acute atherosis in hypertensive pregnancy. American journal of obstetrics and gynecology 123, 164-174
30. Basso, N., and Terragno, N. A. (2001) History about the discovery of the renin-angiotensin system. Hypertension (Dallas, Tex. : 1979) 38, 1246-1249
31. Paul, M., Poyan Mehr, A., and Kreutz, R. (2006) Physiology of Local Renin-Angiotensin Systems. Physiological Reviews 86, 747
32. Poisner, A. M. (1998) The Human Placental Renin–Angiotensin System. Frontiers in neuroendocrinology 19, 232-252
33. Allen, A. M., Zhuo, J., and Mendelsohn, F. A. (1999) Localization of angiotensin AT1 and AT2 receptors. Journal of the American Society of Nephrology : JASN 10 Suppl 11, S23-29
34. Stoll, M., Steckelings, U. M., Paul, M., Bottari, S. P., Metzger, R., and Unger, T. (1995) The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. The Journal of clinical investigation 95, 651-657
35. Morishita, R., Gibbons, G. H., Ellison, K. E., Nakajima, M., Zhang, L., Kaneda, Y., Ogihara, T., and Dzau, V. J. (1993) Single intraluminal delivery of antisense cdc2 kinase and proliferating-cell nuclear antigen oligonucleotides results in chronic inhibition of neointimal hyperplasia. Proceedings of the National Academy of Sciences 90, 8474-8478
36. Paul, M., Poyan Mehr, A., and Kreutz, R. (2006) Physiology of Local Renin-Angiotensin Systems. Physiological Reviews 86, 747-803
37. Paul, M., Poyan Mehr, A., and Kreutz, R. (2006) Physiology of local renin-angiotensin systems. Physiol Rev 86, 747-803
38. Feldt, S., Batenburg, W. W., Mazak, I., Maschke, U., Wellner, M., Kvakan, H., Dechend, R., Fiebeler, A., Burckle, C., Contrepas, A., Jan Danser, A. H., Bader, M., Nguyen, G., Luft, F. C., and Muller, D. N. (2008) Prorenin and Renin-Induced Extracellular Signal-Regulated Kinase 1/2 Activation in Monocytes Is Not Blocked by Aliskiren or the Handle-Region Peptide. Hypertension (Dallas, Tex. : 1979) 51, 682
39. Sowers, J. R., Eggena, P., Kowal, D. K., Simpson, L., Zhu, J.-H., and Barrett, J. D. (1993) Expression of Renin and Angiotensinogen Genes in Preeclamptic and Normal Human Placental Tissue. Hypertension in Pregnancy 12, 163-171
40. Cooper, A. C., Robinson, G., Vinson, G. P., Cheung, W. T., and Broughton Pipkin, F. (1999) The localization and expression of the renin-angiotensin system in the human placenta throughout pregnancy. Placenta 20, 467-474
41. Herr, D., Bekes, I., and Wulff, C. (2013) Local Renin-Angiotensin System in the Reproductive System. Frontiers in Endocrinology 4
42. Schwentner, L., Wöckel, A., Herr, D., and Wulff, C. (2011) Is there a role of the local tissue RAS in the regulation of physiologic and pathophysiologic conditions in the reproductive tract? Journal of the Renin-Angiotensin-Aldosterone System 12, 385-393
43. Kalenga, M. K., De Hertogh, R., Whitebread, S., Vankrieken, L., Thomas, K., and De Gasparo, M. (1991) [Distribution of the concentrations of angiotensin II (A II), A II receptors, hPL, prolactin, and steroids in human fetal membranes]. Revue francaise de gynecologie et d’obstetrique 86, 585-591
44. Williams, P. J., Mistry, H. D., Innes, B. A., Bulmer, J. N., and Broughton Pipkin, F. Expression of AT1R, AT2R and AT4R and Their Roles in Extravillous Trophoblast Invasion in the Human. Placenta 31, 448-455
45. Petit, A., Geoffroy, P., and Belisle, S. (1996) Expression of angiotensin II type-I receptor and phospholipase C-linked G alpha q/11 protein in the human placenta. Journal of the Society for Gynecologic Investigation 3, 316-321
46. Irani, R. A., and Xia, Y. (2008) The Functional Role of the Renin-Angiotensin System in Pregnancy and Preeclampsia. Placenta 29, 763-771
47. Pan, N., Frome, W. L., Dart, R. A., Tewksbury, D., and Luo, J. (2013) Expression of the Renin-Angiotensin System in a Human Placental Cell Line. Clinical Medicine & Research 11, 1-6
48. Xia, Y., Wen, H. Y., and Kellems, R. E. (2002) Angiotensin II inhibits human trophoblast invasion through AT1 receptor activation. The Journal of biological chemistry 277, 24601-24608
49. George, A. J., Thomas, W. G., and Hannan, R. D. (2010) The renin-angiotensin system and cancer: old dog, new tricks. Nature reviews. Cancer 10, 745-759
50. Li, W., Peng, H., Seth, D. M., and Feng, Y. (2012) The Prorenin and (Pro)renin Receptor: New Players in the Brain Renin-Angiotensin System? International Journal of Hypertension 2012, 290635
51. Anton, L., and Brosnihan, K. B. (2008) Systemic and uteroplacental renin–angiotensin system in normal and pre-eclamptic pregnancies. Therapeutic advances in cardiovascular disease 2, 349-362
52. Rodriguez, M., Moreno, J., and Hasbun, J. (2012) RAS in Pregnancy and Preeclampsia and Eclampsia. International Journal of Hypertension 2012, 6
53. Singh, H. J., Rahman, A., Larmie, E. T., and Nila, A. Raised prorenin and renin concentrations in pre-eclamptic placentae when measured after acid activation. Placenta 25, 631-636
54. Seki, H. (2014) The role of the renin–angiotensin system in the pathogenesis of preeclampsia – New insights into the renin–angiotensin system in preeclampsia. Medical Hypotheses 82, 362-367
55. Poisner, A. M. (1995) Regulation of Utero-Placental Prorenin. in Tissue Renin-Angiotensin Systems: Current Concepts of Local Regulators in Reproductive and Endocrine Organs (Mukhopadhyay, A. K., and Raizada, M. K. eds.), Springer US, Boston, MA. pp 411-426
56. Lyall, F., Bulmer, J. N., Duffie, E., Cousins, F., Theriault, A., and Robson, S. C. (2001) Human Trophoblast Invasion and Spiral Artery Transformation : The Role of PECAM-1 in Normal Pregnancy, Preeclampsia, and Fetal Growth Restriction. The American Journal of Pathology 158, 1713-1721
57. Kingdom, J. C. P., and Drewlo, S. (2011) Is heparin a placental anticoagulant in high-risk pregnancies? Blood 118, 4780
58. Chaiworapongsa, T., Chaemsaithong, P., Yeo, L., and Romero, R. (2014) Pre-eclampsia part 1: current understanding of its pathophysiology. Nat Rev Nephrol 10, 466-480
59. Wai Loke, Y., and King, A. (1997) Immunology of human placental implantation: clinical implications of our current understanding. Molecular Medicine Today 3, 153-159
60. Lubel, J., Herath, C., Burrell, L., and Angus, P. (2008) Liver disease and the renin-angiotensin system: Recent discoveries and clinical implications,
61. Delforce, S. J., Lumbers, E. R., and Pringle, K. G. (2017) Regulation of the prorenin – angiotensin system by oxygen and miRNAs; parallels between placentation and tumour development? Placenta 56, 27-33
9.1 Figure Legends
Figure 1. Schematic representation of placental anatomy and development. Placentation involves the invasion of the maternal decidua by extravillous cytotrophoblast cells. Fetal blood vessels are contained within the chorionic villi, which form both floating villi and anchoring villi, and are surrounded by a thin layer of syncytiotrophoblast cells. This layer separates the fetal circulation from the maternal circulation and acts to filter incoming nutrients and substances from the maternal circulation. Invading extravillous cytotrophoblast cells from anchoring villi invade the decidualized endometrium and myometrium (interstitial trophoblasts) and also migrate in a retrograde direction along the maternal spiral arteries (endovascular trophoblasts) transforming them into large diameter conduct vessels of low resistance (56). Waste products are removed from the placental circulation via the maternal vein. Figure adapted from Kingdom, J and Drewlo, S., 2011 (57).
Figure 2. Schematic representation of normal pregnancy spiral artery remodelling (left) and improper spiral artery remodelling featured in preeclampsia (right). In normal pregnancy, invading trophoblast cells extend along spiral arteries through the maternal decidua and one third of the myometrium (58). Both the arterial media and endothelium are destroyed by trophoblasts, this assists the progressive remodelling and dilation of the arteries. Consequently, the delivery of blood and nutrients to the intervillous space is increased (59). In pregnancies affected by preeclampsia, spiral arteries fail to remodel and the result is impaired placentation. Trophoblast invasion is limited to the decidual segment of the spiral artery and does not penetrate the myometrial segment. As a consequence of this shallow invasion, there is a deficiency in the transformation of blood vessels: spiral arteries remain narrow, blood flow is turbulent, and there is an overall reduction in uteroplacental perfusion (58).
Figure adapted from Chaiworapongsa et al. 2014 (58).
Figure 3. The renin-angiotensin system (RAS) cascade. The RAS is depicted here as a linear cascade leading to the generation of angiotensin II (Ang II) (60). Prorenin is activated by binding to the prorenin receptor ((P)RR) and by proteolysis to cleave angiotensin I (Ang I) from angiotensinogen (AGT). Angiotensin converting enzyme (ACE) then converts Ang I to the biologically active Ang II (61). There are two known receptors for Ang II, angiotensin II type-1 receptor (AT1R) and angiotensin II type-2 receptor (AT2R) (60). Ang II binding to AT1R promotes proliferation, angiogenesis, fibrosis, migration and invasion through stimulation of intracellular signalling pathways (61). Conversely, Ang II binding to AT2R opposes AT1R activation. Expanded views of the RAS involve the generation of angiotensin III, angiotensin IV and angiotensin 1-7. These molecular pathways have varying functional effects; however will not be discussed in this review. Figure adapted from Delforce et al. 2017 (61).
10.1 Figures
Figure 1
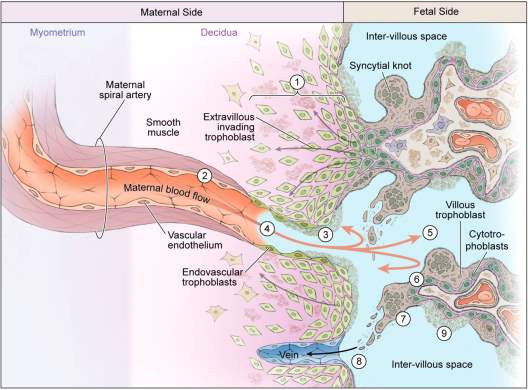
Figure 2
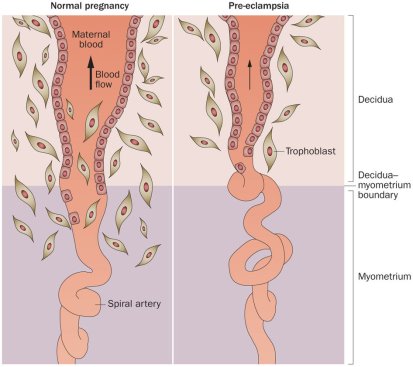
Figure 3
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biomedical Science"
Biomedical Science focuses on how cells, organs and systems function in the human body and underpins much of modern medicine. Biomedical Science applies parts of natural and/or formal sciences to help develop advances in healthcare.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: