Applications of Monoclonal Antibody (mAb) Therapeutics
Info: 8945 words (36 pages) Dissertation
Published: 7th Dec 2021
Tagged: Pharmacology
Antibody Glyco-engineering
Belonging to the immunoglobulin (Ig) superfamily, antibodies are large, Y-shaped glycoproteins produced by plasma cells. As the adaptor molecule in immune system, antibodies can identify and neutralize pathogens such as bacteria, viruses,fungi and chemicals and trigger a cascade of inflammatory mechanisms to eliminate pathogens and resolve infection[1] [2]. In humans, five immunoglobulin classes have been identified (IgG, IgM, IgA, IgE and IgD) that share similar structures composed of Ig domains [3], among which, the most abundant class with longest half-life is IgG comprising 75% of antibodies in serum circulation, IgA and IgM followed up at 15% and 10% individually, while IgE is the least abundant in the serum circulation (
The wide applications of monoclonal antibody (mAb) therapeutics ranging from diagnosis; disease treatments including cancers, autoimmune disease and inflammatory disorder; protein purification and other miscellaneous applications have been expanding continuous progress and technological improvement in the development of mAbs [5]. To date, the majority of recombinant glycoproteins in production from mammalian cell lines such as CHO, NS0 and Sp2/0 are monoclonal antibodies, among which are mainly either chimeric, humanized or human Immunoglobulin Gs (IgGs) [6].
Human IgG consists of four isotypes, IgG1-4, that differ in their ɤ-chain sequences and disulphide bridging patterns, but sharing the same basic structure of IgG molecule [7]. The basic structure of IgG is composed of two light and two heavy chains in covalent and non-covalent association to form three independent protein moieties connected thorough a flexible linker or hinge region (schematic representation in Figure 1)[2]. Two of these moieties are of identical structure and each expresses an antigen specific binding domain (Fab); the third, the fragment crystallizable (Fc) domain, expresses interaction sites for effector functions [2] [8].
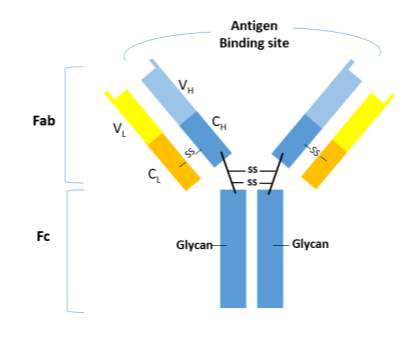
Figure 1 Human IgG structure with complex biantennary glycans attached at Asn297 N-glycosylation site in the CH2 of the Fc region. C: constant domain; V: variable domain; H: heavy chain; L: light chain; S-S: disulfide bond; Fab: Fragment antigen binding domain; Fc: Fragment crystalizable domain.
One of critical post-translational modulations of mAbs is glycosylation, although IgG glycans represent only 3% of the total mass of the molecule, the particular glycoforms are involved in many essential immune effector functions, for example, the ability of an antibody to trigger cell lysis, either through the binding to lymphocyte receptors (FcɤRs, namely FcrRI, FcrRII, FcrRIII) and following a lytic attack from lymphocytes to antibody-targeted cells (referred to as antibody-dependent cellular cytotoxicity, ADCC), or by fixing complement and activation of the complement cascade (complement-dependent cytotoxicity, CDC) via C1q activation or lectin pathway such as mannose binding lectin (MBL) [9] [2]. These carbohydrate moieties are also necessary for maintaining an open conformation of the two heavy chains of IgG and required for maintaining the structural integrity of the FcɤR-binding site [10]. Effector mechanisms mediated through FcɤRs , C1q, MBL and mannose receptor (MR) are severely compromised for aglycosylated IgG-Fc [2]. The deglycosylated IgG antibodies are unable to mediate in vivo triggered inflammatory responses [11]. On the other hand, glycoforms that are not commonly biosynthesized in human would be potentially immunogenic and accelerate the plasmatic clearance of the linked antibody [12].
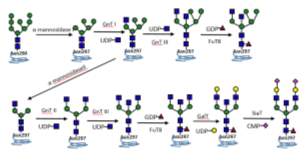
Figure 3 Examples of glycoforms on IgG glycan. A shorthand system of nomenclature for the oligosaccharide uses G0, G1 and G2 for oligosaccharides bearing zero, one or two galactose residues, respectively. When fucose is present G0F, G1F or G2F is used; when bisecting N-acetylglucosamine is present a B is added-for example, G0B or G0BF. The glycoform of the whole molecule is the oligosaccharides present on each heavy chain and may be presented as (G0-G0), (G0-G0F), (G0F-GOF), (GO-G1) and so on.
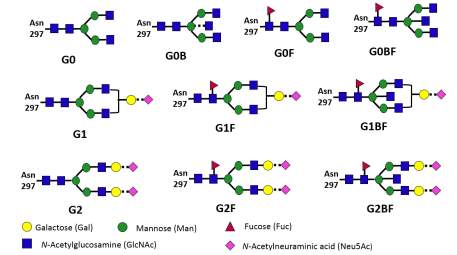
Figure 2 Simplified antibody Fc glycosylation in Golgi apparatus in mammalian cells.
Most human serum IgGs, have complex glycoforms possessing fucosylated bi-antennary glycans with a trimannosyl core capped by an N-acetylgalactosamine (GlcNAc), galactose, and N-acetylneuraminic acid (Neu5Ac) residue on Asparagine 297 in each of the CH2 domains of Fc region (Figure 2). The Fc N-glycans lie within a pocket formed by the 2 CH2 domains of the antibody where they interact with internal amino acid residues [13]. As a consequence of this embedment, the Fc N-glycans are mostly limited to di-antennary complex type with partial galactosylation and low sialylation [14] [2]. Furthermore, the majority of human IgG Fc glycans are highly fuocsylated (>92%), containing G0F (~35%) G1F (~35%), and G2F (~16%) for the fucosylated glycans [7]. A small proportion of human IgG Fc glycans contains a bisecting GlcNAc (>11%), as shown in Fig.2. Only 5~10% of IgG Fc glycoforms are mono-sialylated and less than 1% of IgG Fc glycoforms possesses two sialic acids [7] [15] [16] [17] [4]. Whereas, recombinant IgGs expressed in CHO and mouse myeloma NS0 and Sp2/0 cells generally less galactosylated, tending to contain higher level of G0 glycans [6]. Some of these glycoforms, like galactosylation and sialylation, are desirable for decreasing immunogenicity, and others, such as afucosylation, bisecting GlcNAc residues, and high-mannose glycans, enhancing ADCC activity [18] [19].
Given the multifunctional roles of Fc glycans on antibody therapeutics, in this review, we will focus on the contribution of glycosylation strategies on IgG antibody engineering and mainly discuss the current achievements of sialylation, galactosylation, defucosylation, GnT-III overexpression and GnT-I knockout on IgG Fc glycoengineering, then we also cover the heterogeneity of glycans and emerging technologies for producing homogenous glycans on antibodies.
1. Sialylation
For most therapeutic glycoproteins except for antibodies, the terminal sialic acids on glycans have a clear and critical role for proteins’ circulatory lifespan and efficacy. Serving as a biological mask, the distal sialic acid can shield Gal residues that when exposed prompt a fast removal of the protein from blood circulation due to endocytosis-mediated uptake by asialoglycoprotein receptors on the hepatocytes [20] [21] [22]. Therefore, in mammalian cells, it is generally desirable to maximize the distal sialic acid content of a glycoprotein in order to ensure its quality and consistency as an effective therapeutic [23]. However, for antibody glycosylation, the IgG Fc sialylation has emerged as an important but controversial concept— increase IgG Fc sialylation enhances the anti-inflammatory activity of antibodies [24] but impair CDC [25]; while decrease Fc sialylation can facilitate FcγR receptors binding on natural killer (NK) cells and lower affinity binding to cell-surface antigen [26], as well as increase ADCC activity [14] [27].
It has been reported that intravenous immunoglobulin (IVIG) with sialylation on the glycan of Fc portion exhibits superior anti-inflammatory efficacy over asialylated IVIG [27]. Especially, the presence of α2,6-linked sialic acid residues play a critical role in inducing IVIG-elicited anti-inflammatory activity [28] [29] [26]. Despite that half-life of IVIG may not be significant improved due to the generally low sialylation content of IgGs, increasing recombinant IgG sialylation containing α2,6-linked sialic acid would enhance its anti-inflammatory activity. On the other hand, human proteins have a2,3- and a2,6- linkages, predominantly in α2,6-linkage [30], whereas CHO cells have incomplete sialylation with exclusively α2,3-linked sialic acid. Therapeutic proteins possessing both a2,3- and a2,6-sialic acid linkages are more “human-like” and are less likely to be immunogenic.
The sialylation of antibody is controlled primarily by the interaction of IgG structure local to the carbohydrate and the accessibility of sialyltransferase to sialylate the nascent polypeptide[31]. In human, 10-15% of the circulating IgG1 are sialylated, carrying mostly complex di-antennary glycans with two Gal and one sialic acid, whereas the mAbs produced in CHO and mouse myeloma NS0 and Sp2/0 cells typically possesses N-glycans with
In addition to the amino acid substitution approach, introduction of heterogeneous a2,6-ST in recombinant IgG producing CHO cell line would further increase the sialylation content [38] [30] . For instance, the F243A replacement resulted in 53% of a2,3 sialylation on the recombinant IgG3 produced in CHO, and following transfection of rat ST6GAL1 into this line, the IgG-Fc-linked glycans were 60% sialylated [38].While overexpression of exogenous ST6GAL1 in CHO could only moderately enhance a2,6-sialylated glycans, and the a2,6 sialylation content never exceed a2,3 sialylation content [39] [30]. Recently, the report on genomic sequence of the CHO-K1 cell line revealed that ST6GAL1 gene is retained in CHO genome, but its expression is severely suppressed and regulated [40]. And introduction of CHO ST6GAL1gene into the CHO cell line could also serve as an option to enhance the a2,6 sialylation level of recombinant proteins in CHO[41]. Moreover, co-overexpression of β1,4-GalT and a2,6-ST would achieve a glycoprofile dominated by a2,6-sialylation in recombinant antibody production[14].
On the other hand, in order to optimize ADCC, several approaches have been attempted to reduce IgG Fc sialylation. For instance, expression of exogenous sialidase A gene in mammalian cells resulted in the expression of soluble enzyme that was capable of removing sialic acid from antibodies secreted into the medium. And ADCC assay showed that the two Abs expressed in sialidase A-transfected cells had greater than 10-fold improved potency compared to the control mock-transfected Abs [27]. Using zinc-finger nucleases to inactive CMP-sialic acid transporter gene resulted in a mutant CHO cell line lacking functional CMP-sialic acid transporter and the removal of sialic acid from IgG1 enhanced ADCC [42] .
2.2 Galactosylation
Given the low sialylation content of Fc glycans in both human serum IgG and recombinant IgGs, the Fc glycans contain 0,1, or 2 terminal Gal residues (G0, G1 or G2) in their antennae [43], among which the predominant glycoform is G0F with terminal N-acetylglucosamine sugar residues. The recombinant IgGs expressed in CHO cells are generally less galactosylated and tend to contain higher levels of G0 glycans compared with IgGs produced in mouse myeloma cell lines [6]. The terminal galactosylation of IgG has been reported to affect CDC, but not impact ADCC activity [44] [45] [46] or the antibody binding to antigen [6]. Removal of terminal galactose residues from a chimeric mouse-human IgG1 antibody was shown to reduce CDC but to be without effect on FCrR-mediated functions [47]. For Rituxan, an increase in terminal Gal content can increase CDC activity as a result of increased antibody binding to C1q [48]. Similarly, the G1 glycoform of rituximab triggered a CDC response twice as large as that triggered by the G0 glycoform [18].Moreover, antibody galactosylation, on the other hand, was demonstrated to impact CDC complement activation by the lectin pathway-mannose binding lectin (MBL), but not by classic Fc receptor-mediated functions [49] [50].
While another consideration when looking at the functional influence of terminal galactose is that sialic acid is added on the penultimate galactose of glycan by sialyltransferases, as depicted in Fig. 1 [51]. Thus, the enhancement of sialylation is generally followed with an increase of galactosylation content. IgG 1 Abs lacking galactose have shown aberrant activity that might contribute to inflammatory disorders [52] [53]. And a recent report show that that highly galactosyated IgG1 mediates anti-inflammatory activity by facilitating the association of the inhibitory receptor FcRIIB and a C-type lecin-like receptor dectin-1[4] [54]. However, the effect of galacsosylation on eliciting anti-inflammation appears to be subclass specific and needs further exploration , since highly galactosylated IgG2 failed to initiate such inhibition [4].
2.2 Defucosylation
Numerous studies have shown that the core-fucose residue of the CH2-associated N-linked oligosaccharides plays a critical role in regulating the magnitude of antibody effector function ADCC [55] [56] [57] [58] [59]. Compared to the fucosylated antibodies from wide-type CHO cells, defucosylated antibodies exhibited 100-fold higher ADCC in vitro [44]. The core fucose is bound through an α-1,6 linkage to the innermost GlcNAc residue on an N-oligosaccharide, which could hinder Fc binding to lymphocyte receptors[60] [58]. It has been demonstrated that the antibody ADCC enhancement due to fucose elimination is attributed to a subtle conformational alteration in a limited region of IgG1-Fc, resulting in higher Fc binding to gamma receptor IIIa (Fc FcɤRIIIa) on lymphocytes[59, 61] [62]. Compared to amino acid mutations in the antibody Fc region, enhanced binding affinity for FcɤRIIIa of fucose-negative antibody alone is sufficient to induce maximal ADCC [63] .
Moreover, antibodies with Fc regions containing nonfucosylated N-glycans exhibit dramatically enhanced ADCC without any detectable change in CDC or antigen binding capability [58] [64]. Only the lack of fucose, but not sialic acid or galactose is responsible to bind to FcrRIIIa and in the recruitment of mononuclear cells (primarily NK cells) as effector cells for ADCC[55, 65, 66]. However, studies on polymorphonuclear cells (PMNs) were found that high-fucosylated antibodies were preferentially more effective in triggering PMN, which mediated tumor cell lysis via FcrRII[66]. Thus, the impact of Fc fusosylation on ADCC still need further exploration and may be critically dependent on the specific effector cell type recruited. Given the majority of monoclonal antibodies in the market are targeted to mediate cytotoxicity of NK effector cells, we will focus on the defucosylation of antibodies.
2.2.1 Inactivation of α1,6 fucosyltransferase—Disruption of FUT8 gene
Inactivation of the α1,6 fucosyltransferase has been extensively studied and still the most efficient modification to prevent the addition of fucose to the core of the complex N-glycan (Figure 1). In mammals, the FUT8 gene encodes α-1,6 fucosyltransferase in the medial Golgi cisternae that catalyzes the transfer of fucose residues from GDP-fucose to the innermost GlcNAc of the tri-mannosyl core structure via the α1,6 linkage[67]. In order to generate non-fucosylated antibody, disruption of FUT8 gene has been studied via various approaches as discussed below [68] [58] [69] [70].
Traditionally, homologous recombinantion such as antisense cDNA, shRNA or transgenic overexpression methods was used to reduce or remove fucosylation [71]. The introduction of small interfering RNAs (siRNAs) against FUT8 gene reduced FUT8 mRNA expression to 20% in CHO/DG44 cells and combined with Lens culinaris agglutinin (LCA) selection yielded 60% antibody defucosylation with over 100-fold higher ADCC [68]. Disrupt both FUT8 alleles in CHO/DG44 cell line by sequential homologous recombination produced completely defucosylated recombinant antibodies, FUT8-/- produced chimeric anti-CD20 IgG1 showed the same level of antigen-binding activity and CDC as the FUT8+/+-produced [58]. However, the time-consuming homologous recombination strategies either cannot produce completely nonfucosylated antibodies or the complete knockout of FUT8 is reversible and the difficulty of recapitulating the knockout in protein-production cell lines has prevented the widespread adoption of homologous recombination FUT8-/- cells as hosts for antibody production [72] [71] [58]. To improve the efficiency of disrupting FUT8 gene, technologies such as zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) were developed and reported [72] [73]. Using zinc-finger nucleases (ZFNs) to disrupt FUT8 gene in a region encoding the catalytic core of the enzyme produced FUT8-/- CHO cells in 3 weeks at a frequency of 5% in the absence of any selection, and these lines produced antibodies completely lacking core fucosylation but having an otherwise normal glycosylation pattern without adverse phenotypic effects [72]. Besides, Cristea et al. demonstrated knockout of FUT8 gene via the simultaneous TALE nuclease-mediate integration of an antibody cassette [73]. Composed of programmable, sequence-specific DNA-binding modules linked to a nonspecific DNA cleavage domain, ZFNs and TALENs enable a broad range of genetic modulations by inducing DNA double-strand breaks that stimulate error-prone nonhomologous end joining (NHEJ) or homology-directed repair (HDR) at specific genomic locations [74] [75]. Nevertheless, NHEJ could be harnessed to mediate gene functional disruptions, as indels occurring in a coding region could lead to frameshift mutations and protein function loss [76] [77].
More recently, distinct from the site-sepcific ZFNS and TALENS, the CRISPR (clustered regularly interspaced short palindromic repeat)/Cas9 (CRISPR-associated protein 9) system has emerged as a potentially facile and efficient alternative for inducing targeted genetic alterations [75], which was derived from the microbial adaptive immune system [78], and applied to diverse species such as plants, animals, bacteria, and yeast [79], as well as in CHO cells [80] [77] . This system required only Cas9 and single guide RNA (gRNA) [81], giving it several advantages including ease of customization, higher targeting efficiency, and multiplexed genome editing capability [82]. Sun et al. reported functional FUT8 disruptive clones (FUT8-/-) in CHO cells using CRISPR/Cas9 were obtained within 3 weeks at indel frequencies ranging from 9 to 25%, which was enhanced to 52% with LCA selection and the derived FUT8-/- clone had the ability to produce defucosylated therapeutic mAb with no detrimental effects on cell growth, viability or product quality [77]. Moreover, Grav et al. employed CRISPR/Cas 9 targeted FUT8, BaK and BAX triple knockout simultaneously and created a cell line produced afucoylated antibody with improved protein productivity [83].
2.2.2 Inhibit the supply of nucleotide sugar substrate for fucosylation
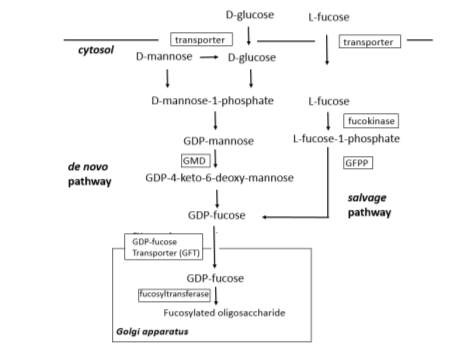
Figure 4 The pathways of oligosaccharide fucosylation in mammalian cell lines.
An alternative approach for producing fucose-negative antibody is to knockout enzymes in the making of GDP-fucose, the donor for fucosyltransferase in the Golgi apparatus. The advantage of this approach is that it targets all fucosyltransferases, not just FUT8 [71]. In mammalian cells, GDP-fucose is synthesized by two distinct pathways as illustrated in Figure4. The de novo pathway is characterized by conversion of GDP-mannose to GDP-4-keto-6-deoxymannose by GDP-mannose 4,6-dehydratase (GMD). This keto intermediate is then converted to GDP-fucose by GDP-4-keto-6-deoxymannose-3,5-epimerase-4-reductase (GMER). The salvage pathway can directly utilizes fucose that is transported into the cytosol from an extracellular origin or fucose that is liberated from catabolism of fucosylated glycans in the lysosome and then transported into the cytosol [84]. The salvage pathway is enabled by fucokinase and GDP-fucose pyrophospohorylase (GFPP) [84]. Subsequently, GDP-fucose synthesized by these two pathways is transported into the lumen of the Golgi apparatus by GDP-fucose transporter (GFT) [85].
Loss-of-function analyses using siRNA revealed that three key genes involved in oligosaccharide fucosylation, i.e. a 1,6-fucosyltransferase (FUT8), GDP-mannose 4,6-dehydratase (GMD), and GDP-fucose transporter (GFT), and single-gene knockdown of each target was insufficient to completely defucosylate the products in antibody-producing cell lines [86]. For GFT, introduction of a CHO GFT siRNA expression plasmid into human antithrombin III (AT-III)-producing cell line showed approximately 75% reduction of Golgi-GDP fucose, resulting in 10-40% defucosylation in AT-III [85]. Similarly, Kanda et al. reported expression levels of GFT mRNA could be reduced more than 90% in GFT siRNA-expressed antibody-producing CHO cells, but only 20-40% reduction of the recombinant IgG Fc oligosaccharide fucosylation was observed [87]. The positive relation between the Fc oligosaccharide fucosylation and the mRNA expression for both GMD and FUT8, but not for the GDP-fucose transporter suggested that GMD and FUT8 share no redundancy, and are independently responsible for α-1,6 fucosylation, without complementing one another [87] .
Alternatively, a variant of Pro- CHO 5 cell, Lec 13, has been isolated using lens culinaris agglutinin (LCA) selection, with a loss of GMD activity resulting in only 10% of fucose residing on the glycan chain compared to the parental cell line [88]. But Lec13 is unsuitable for the serum-free fed-batch production of fucose-negative antibody because the remaining GMD activity in Lec13 accumulates intracellular GDP-fucose via the de nove pathway and gradually increases the ratio of fucosylated products with extended culture [89]. Moreover, although GMD knockout CHO/DG44 cells were able to be devoid of intracellular GDP-fucose and to produce completely non-fucosylated antibodies, the fucosyaltion could be recovered through the salvage pathway upon the addition of L-fucose into the culture medium [87]. But double knockdown of FUT8 and GMD using short hairpin siRNA tandem expression vector could rapidly converted antibody-producing CHO cells to fully non-fucosylated antibody producers in serum-free fed-batch culture [86].
2.3 Overexpression of GnT-III
Furthermore, a different and complementary approach to improve antibody’s biological activity would be to overexpress β 1,4-N-acetylglucosaminyltransferase III (GnT-III) enzyme to produce antibodies enriched in bisecting oligosaccharides. GnT-III is a Golgi-localized enzyme and catalyzes the addition of an N-acetylglucosamine (GlcNAC) residue to a bisecting position of the N-linked oligosaccharide, as shown in figure 1 [90]. Overexpression of GnT-III gene in a CHO cell line expressing an antioneuroblastoma IgG1 resulted in greater ADCC activity, because GnT-III overexpression leads to increase non-fucosylated and hybrid oligosaccharides content and further enhance binding affinity for FcɤRs [91] [92]. Overexpression of GnT-III in a CHO cell line producing anti CD-20 antibody resulted in a 20-fold lower antibody dosage to achieve the same ADCC effect induced by the antibody produced from the parental CHO cell line [93]. Besides the GnT-III localization also has been studied. By fusing the catalytic domain of GnT-III to the localization domain (cytoplasmic, transfermembrane, and stem region) of other Golgi-resident enzymes of N-glycosylation pathway in HEK293-EBNA cells, the chimeric GnT-III can compete even more efficiently against the endogenous core a1,6-fucosyltransferase, leading to mainly bisected non-fucosylated hybrid glycans (hybrid-rich) [91]. Golgi Man II yields the precursor for all biantennary, complex N-glycans as illustrated in Figrue 1. Thus, co-expressing GnT-III and Man II led to higher proportions of bisected non-fucosylated complex glycans (complex-rich) [91]. While both glycovariants featured strongly increased ADCC activity compared to the wild-type antibody, the hybrid-rich antibody showed reduced CDC compared to complex-rich or wild-type counterparts [92] [94]. It is worth noting that the effect of bisecting GlcNAc of the IgG1 on enhancing ADCC is modest when compared to that of defucosylated IgG1, therefore, the lack of fucosylation has the most critical role in enhancement of ADCC[93] [95] [56].
2.4 GnT-I knockout to generate high-mannose glycans
Different from complex and hybrid N-glycan types, high mannose glycans (Man5-9GlcNAc2) are characterized by unsubstituted terminal mannose sugars of five to nine residues attached to the GlcNAc2 core [96]. They are typically non-natural glycoforms and accounts for below 5% in recombinant therapeutic antibody glycoprofile, but they have a major impact on in vivo therapeutic activity— enhancing ADCC , decreasing CDC and having faster clearance rate [97] [98] [99, 100]. Moreover, high-mannose glycans has been widely applied to generate minimally glycosylated glycoproteins for X-ray crystallography [101].
Golgi N-acetylglycosaminyltransferase I (GnT-I, also called Mgat1) is a type II transmembrane enzyme that plays a key role in the biosynthesis of N-linked glycans [102]. As shown in Figure 1, it specifically transfers GlcNAc from UDP-GlcNAc onto the oligomannose core to initiate complex and hybrid N-linked carbohydrate synthesis in the medial Golgi [102]. Thus, methods for increasing high mannose levels focus on inhibiting glycosylation in or before the medial-Golgi, mainly knockout and knockdown GnT-I gene.
For high mannose glycans, one of the most widely used and numerously studied cell lines is the Lec1 CHO mutant cell line [19]. Owing to their defect in GnT-I, Lec1 cells synthesize only oligomannose-type N-glcyans [103] [102, 104]. Over the years, there are two discrepencies about the Lec 1 cells, one concerns the unpredictable high mannose oligosaccharides (Man5-Man9) distribution in Lec 1 cells[105] [62] [100], another focuses on the uncertain existence of core α1,6 fucosylaiton in Lec 1 cell line [106] [100] [107], which prohibits its extensive application in industry. Recently, new high mannose cell lines have been explored and established using novel genetic techniques to study GnT-I and its role in glycosylation. Sealover et al. reported the use of zinc-finger nuclease (ZFN) genome editing technology to disrupt GnT-I gene in CHO, generating cell lines that produce recombinant proteins predominantly bearing Man5 glycans [108]. They successfully isolated GnT-I knockout clones without lectin enrichment by screening a manageable number of clones [108]. ZFN-based genome editing eliminates potential regulatory concerns associated with random chemical mutagenesis. ZFN technology succeeds in generating 5 clones out of 90 (5.6%) containing deletions in the GnT-I gene and all five GnT-I-deficient cell lines demonstrated similar growth and productivity characteristics to the wild-type CHO-K1 host cell line [108]. Unlike protein-based targeting ZFN method, Lee et al. later demonstrated efficient targeted gene integration into site-specific loci in CHO cell through CRISPR/Cas9 genome editing system with compatible donor plasmid harboring short-homology arms, GOI, and a fluorescent marker gene as an indicator of random integration[109]. By using CRISPR/Cas9 and homology-directed DNA repair (HDR) mediated targeted integration system, GnT-I locus was completely disrupted in most target clonal cells (20 out of 25 clones), which showed a relative GnT-I copy number of close to 0 compared with the GnT-I in CHO-S wild type cells[109].
2. Heterogeneity of glycans
Another issue in the N-glycans of monoclonal antibodies is heterogeneous glycoforms, which cause problems for antibody purification and product consistency and reproducibility and can lead to variable therapeutic efficacy. For instance, this diversity can adversely affect drug potency and pharmacokinetics [91] [110]. However, N-glycans are often crucial for protein folding, so these difficulties cannot be overcome by removing N-glycosylation sites [111]. Heterogeneity is attributed to the
To address this issue, Meuris et al. introudced a new strategy (GlycoDelete) of shortcutting N-glycosylation to present simple and homogenous glycoproteins expressed in HEK293S cells [111]. Briefly, the authors inactivated N-acetylglucosaminyltransferase I (GnT-I) to produce high-mannose glycans. Then they overexpressed an endo-N-acetylglucsoaminidase targeted to the trans-Golgi, followed by Concanavalin A (ConA) lectin selection. With the following actions galatransferases and sialytransferses in the Golgi, GlycoDelete cells produce proteins with the Gal-GlcNAc disaccharide or its α-2,3-sialylated trisaccharide derivative and some of the monsaccharide intermediate [111] . To assess the potential of these GlycoDelete glycans and their influence on therapeutic proteins, human GM-CSF and an anti-CD20 antibody were produced in HEK293s and HEK293sGlycoDelete cells [111]. For hGM-CSF no significant influence of the GlycoDelete sugars on the activity of the protein has been observed, GlycoDelete anti-CD20 on the other hand has a significantly reduced FcɤR affinity and an increased circulation time in mice compared to HEK293s produced anti-CD20 [111] [112].
Moreover, in order to provide homogenous glycoform, Zhang et al. conducted a comprehensive Zinc-finger nucleases knockout screen of 19 glycosyltransferase genes and identified the key genes that control decisive steps in N-glycosylation in CHO [113]. The author reported stacking knockout of GnT-IV-A/GnT-IV-B/GnT-V produced almost homogenous biantennary N-glycans [113]. Subsequently, introduction of ST6Gal-I in CHO ST3Gal4/6 knockout cells produced normal range of N-glycan with only a2,6-sialylation, when combined with GnT-IV-A/GnT-IV-B/GnT-V knockout, homogenous biantennary N-glycan capped by a2,6-sialylation were generated [113].
References
1. Buss, N.A., et al., Monoclonal antibody therapeutics: history and future. Curr Opin Pharmacol, 2012. 12(5): p. 615-22.
2. Jefferis, R., Glycosylation of natural and recombinant antibody molecules. Adv Exp Med Biol, 2005. 564: p. 143-8.
3. Amzel, L.M. and R.J. Poljak, Three-dimensional structure of immunoglobulins. Annu Rev Biochem, 1979. 48: p. 961-97.
4. Shade, K.-T. and R. Anthony, Antibody Glycosylation and Inflammation. Antibodies, 2013. 2(3): p. 392.
5. Niwa, R. and M. Satoh, The Current Status and Prospects of Antibody Engineering for Therapeutic Use: Focus on Glycoengineering Technology. Journal of Pharmaceutical Sciences, 2015. 104(3): p. 930-941.
6. Raju, T.S. and R.E. Jordan, Galactosylation variations in marketed therapeutic antibodies. MAbs, 2012. 4(3): p. 385-91.
7. Arnold, J.N., et al., The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol, 2007. 25: p. 21-50.
8. Buss, N.A.P.S., et al., Monoclonal antibody therapeutics: history and future. Current Opinion in Pharmacology, 2012. 12(5): p. 615-622.
9. Beck, A., et al., Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol, 2010. 10(5): p. 345-52.
10. Yamaguchi, Y., et al., Glycoform-dependent conformational alteration of the Fc region of human immunoglobulin G1 as revealed by NMR spectroscopy. Biochim Biophys Acta, 2006. 1760(4): p. 693-700.
11. Nimmerjahn, F. and J.V. Ravetch, Fcgamma receptors: old friends and new family members. Immunity, 2006. 24(1): p. 19-28.
12. Beck, A., et al., Characterization of therapeutic antibodies and related products. Anal Chem, 2013. 85(2): p. 715-36.
13. Wormald, M.R., et al., Variations in oligosaccharide-protein interactions in immunoglobulin G determine the site-specific glycosylation profiles and modulate the dynamic motion of the Fc oligosaccharides. Biochemistry, 1997. 36(6): p. 1370-80.
14. Raymond, C., et al., Production of alpha 2,6-sialylated IgG1 in CHO cells. Mabs, 2015. 7(3): p. 571-583.
15. Zauner, G., et al., Glycoproteomic analysis of antibodies. Mol Cell Proteomics, 2013. 12(4): p. 856-65.
16. Stadlmann, J., et al., A close look at human IgG sialylation and subclass distribution after lectin fractionation. Proteomics, 2009. 9(17): p. 4143-53.
17. Wuhrer, M., et al., Glycosylation profiling of immunoglobulin G (IgG) subclasses from human serum. Proteomics, 2007. 7(22): p. 4070-81.
18. Jefferis, R., Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov, 2009. 8(3): p. 226-234.
19. Shi, H.H. and C.T. Goudar, Recent advances in the understanding of biological implications and modulation methodologies of monoclonal antibody N-linked high mannose glycans. Biotechnology and Bioengineering, 2014. 111(10): p. 1907-1919.
20. Ashwell, G. and J. Harford, Carbohydrate-specific receptors of the liver. Annu Rev Biochem, 1982. 51: p. 531-54.
21. Cole, E.S., et al., Invivo Clearance of Tissue Plasminogen-Activator – the Complex Role of Sites of Glycosylation and Level of Sialylation. Fibrinolysis, 1993. 7(1): p. 15-22.
22. Wang, Q., et al., Strategies for Engineering Protein N-Glycosylation Pathways in Mammalian Cells. Methods Mol Biol, 2015. 1321: p. 287-305.
23. !!! INVALID CITATION !!! [13].
24. Kaneko, Y., F. Nimmerjahn, and J.V. Ravetch, Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science, 2006. 313(5787): p. 670-3.
25. Quast, I., et al., Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J Clin Invest, 2015. 125(11): p. 4160-70.
26. Scallon, B.J., et al., Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Molecular Immunology, 2007. 44(7): p. 1524-1534.
27. Naso, M.F., et al., Engineering host cell lines to reduce terminal sialylation of secreted antibodies. MAbs, 2010. 2(5): p. 519-27.
28. Anthony, R.M., et al., Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science, 2008. 320(5874): p. 373-6.
29. Anthony, R.M. and J.V. Ravetch, A novel role for the IgG Fc glycan: the anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol, 2010. 30 Suppl 1: p. S9-14.
30. Lin, N., et al., Chinese hamster ovary (CHO) host cell engineering to increase sialylation of recombinant therapeutic proteins by modulating sialyltransferase expression. Biotechnol Prog, 2015. 31(2): p. 334-46.
31. Lund, J., et al., Multiple interactions of IgG with its core oligosaccharide can modulate recognition by complement and human Fc gamma receptor I and influence the synthesis of its oligosaccharide chains. J Immunol, 1996. 157(11): p. 4963-9.
32. Raymond, C., et al., Production of IgGs with a human-like sialylation in CHO cells. BMC Proceedings, 2015. 9(9): p. 1-2.
33. Mimura, Y., et al., Glycosylation of Therapeutic IgGs, in Therapeutic Monoclonal Antibodies. 2009, John Wiley & Sons, Inc. p. 67-89.
34. Jefferis, R. and J. Lund, Glycosylation of antibody molecules: structural and functional significance. Chem Immunol, 1997. 65: p. 111-28.
35. Mimura, Y., et al., Enhanced sialylation of a human chimeric IgG1 variant produced in human and rodent cell lines. J Immunol Methods, 2016. 428: p. 30-6.
36. Subedi, Ganesh P. and Adam W. Barb, The Structural Role of Antibody N-Glycosylation in Receptor Interactions. Structure. 23(9): p. 1573-1583.
37. Yu, X., et al., Engineering Hydrophobic Protein–Carbohydrate Interactions to Fine-Tune Monoclonal Antibodies. Journal of the American Chemical Society, 2013. 135(26): p. 9723-9732.
38. Jassal, R., et al., Sialylation of Human IgG-Fc Carbohydrate by Transfected Rat α2,6-Sialyltransferase. Biochemical and Biophysical Research Communications, 2001. 286(2): p. 243-249.
39. Jassal, R., et al., Sialylation of human IgG-Fc carbohydrate by transfected rat alpha2,6-sialyltransferase. Biochem Biophys Res Commun, 2001. 286(2): p. 243-9.
40. Xu, X., et al., The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotech, 2011. 29(8): p. 735-741.
41. Onitsuka, M., et al., Enhancement of sialylation on humanized IgG-like bispecific antibody by overexpression of alpha2,6-sialyltransferase derived from Chinese hamster ovary cells. Appl Microbiol Biotechnol, 2012. 94(1): p. 69-80.
42. Haryadi, R., et al., CHO-gmt5, a novel CHO glycosylation mutant for producing afucosylated and asialylated recombinant antibodies. Bioengineered, 2013. 4(2): p. 90-4.
43. Raju, T.S., Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol, 2008. 20(4): p. 471-8.
44. Peipp, M., et al., Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood, 2008. 112(6): p. 2390-9.
45. Shinkawa, T., et al., The Absence of Fucose but Not the Presence of Galactose or Bisecting N-Acetylglucosamine of Human IgG1 Complex-type Oligosaccharides Shows the Critical Role of Enhancing Antibody-dependent Cellular Cytotoxicity. Journal of Biological Chemistry, 2003. 278(5): p. 3466-3473.
46. Hodoniczky, J., Y.Z. Zheng, and D.C. James, Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol Prog, 2005. 21(6): p. 1644-52.
47. Boyd, P.N., A.C. Lines, and A.K. Patel, The effect of the removal of sialic acid, galactose and total carbohydrate on the functional activity of Campath-1H. Molecular Immunology, 1995. 32(17–18): p. 1311-1318.
48. Hodoniczky, J., Y.Z. Zheng, and D.C. James, Control of Recombinant Monoclonal Antibody Effector Functions by Fc N-Glycan Remodeling in Vitro. Biotechnology Progress, 2005. 21(6): p. 1644-1652.
49. Malhotra, R., et al., Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med, 1995. 1(3): p. 237-43.
50. Nimmerjahn, F., R.M. Anthony, and J.V. Ravetch, Agalactosylated IgG antibodies depend on cellular Fc receptors for in vivo activity. Proc Natl Acad Sci U S A, 2007. 104(20): p. 8433-7.
51. Vidarsson, G., G. Dekkers, and T. Rispens, IgG subclasses and allotypes: from structure to effector functions. Front Immunol, 2014. 5: p. 520.
52. Rademacher, T.W., et al., The role of IgG glycoforms in the pathogenesis of rheumatoid arthritis. Springer Seminars in Immunopathology, 1988. 10(2): p. 231-249.
53. Axford, J.S., et al., Changes in normal glycosylation mechanisms in autoimmune rheumatic disease. J Clin Invest, 1992. 89(3): p. 1021-31.
54. Karsten, C.M., et al., Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med, 2012. 18(9): p. 1401-6.
55. Shields, R.L., et al., Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem, 2002. 277(30): p. 26733-40.
56. Shinkawa, T., et al., The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem, 2003. 278(5): p. 3466-73.
57. Satoh, M., S. Iida, and K. Shitara, Non-fucosylated therapeutic antibodies as next-generation therapeutic antibodies. Expert Opin Biol Ther, 2006. 6(11): p. 1161-73.
58. Yamane-Ohnuki, N., et al., Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng, 2004. 87(5): p. 614-22.
59. Iida, S., et al., Nonfucosylated therapeutic IgG1 antibody can evade the inhibitory effect of serum immunoglobulin G on antibody-dependent cellular cytotoxicity through its high binding to FcgammaRIIIa. Clin Cancer Res, 2006. 12(9): p. 2879-87.
60. Nagae, M. and Y. Yamaguchi, Function and 3D Structure of the N-Glycans on Glycoproteins. International Journal of Molecular Sciences, 2012. 13(7): p. 8398-8429.
61. Matsumiya, S., et al., Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J Mol Biol, 2007. 368(3): p. 767-79.
62. Kanda, Y., et al., Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N-linked Fc oligosaccharides: the high-mannose, hybrid, and complex types. Glycobiology, 2007. 17(1): p. 104-18.
63. Masuda, K., et al., Enhanced binding affinity for FcgammaRIIIa of fucose-negative antibody is sufficient to induce maximal antibody-dependent cellular cytotoxicity. Mol Immunol, 2007. 44(12): p. 3122-31.
64. Yamane-Ohnuki, N. and M. Satoh, Production of therapeutic antibodies with controlled fucosylation. mAbs, 2009. 1(3): p. 230-236.
65. Okazaki, A., et al., Fucose Depletion from Human IgG1 Oligosaccharide Enhances Binding Enthalpy and Association Rate Between IgG1 and FcγRIIIa. Journal of Molecular Biology, 2004. 336(5): p. 1239-1249.
66. Peipp, M., et al., Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood, 2008. 112(6): p. 2390-2399.
67. Miyoshi, E., et al., The alpha1-6-fucosyltransferase gene and its biological significance. Biochim Biophys Acta, 1999. 1473(1): p. 9-20.
68. Mori, K., et al., Engineering Chinese hamster ovary cells to maximize effector function of produced antibodies using FUT8 siRNA. Biotechnol Bioeng, 2004. 88(7): p. 901-8.
69. Ma, B., J.L. Simala-Grant, and D.E. Taylor, Fucosylation in prokaryotes and eukaryotes. Glycobiology, 2006. 16(12): p. 158r-184r.
70. Mori, K., et al., Non-fucosylated therapeutic antibodies: the next generation of therapeutic antibodies. Cytotechnology, 2007. 55(2-3): p. 109-14.
71. Stanley, P., Chinese hamster ovary mutants for glycosylation engineering of biopharmaceuticals. Pharmaceutical Bioprocessing, 2014. 2(5): p. 359-361.
72. Malphettes, L., et al., Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnol Bioeng, 2010. 106(5): p. 774-83.
73. Cristea, S., et al., In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnology and Bioengineering, 2013. 110(3): p. 871-880.
74. Urnov, F.D., et al., Genome editing with engineered zinc finger nucleases. Nat Rev Genet, 2010. 11(9): p. 636-46.
75. Gaj, T., C.A. Gersbach, and C.F. Barbas, ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends in biotechnology, 2013. 31(7): p. 397-405.
76. Perez, E.E., et al., Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotech, 2008. 26(7): p. 808-816.
77. Sun, T., et al., Functional knockout of FUT8 in Chinese hamster ovary cells using CRISPR/Cas9 to produce a defucosylated antibody. Engineering in Life Sciences, 2015. 15(6): p. 660-666.
78. Barrangou, R., et al., CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science, 2007. 315(5819): p. 1709-1712.
79. DiCarlo, J.E., et al., Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Research, 2013.
80. Ronda, C., et al., Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnology and Bioengineering, 2014. 111(8): p. 1604-1616.
81. Mali, P., et al., RNA-Guided Human Genome Engineering via Cas9. Science, 2013. 339(6121): p. 823-826.
82. Ran, F.A., et al., Genome engineering using the CRISPR-Cas9 system. Nat. Protocols, 2013. 8(11): p. 2281-2308.
83. Grav, L.M., et al., One-step generation of triple knockout CHO cell lines using CRISPR/Cas9 and fluorescent enrichment. Biotechnology Journal, 2015. 10(9): p. 1446-1456.
84. Becker, D.J. and J.B. Lowe, Fucose: biosynthesis and biological function in mammals. Glycobiology, 2003. 13(7): p. 41R-53R.
85. Omasa, T., et al., Decrease in antithrombin III fucosylation by expressing GDP-fucose transporter siRNA in Chinese hamster ovary cells. J Biosci Bioeng, 2008. 106(2): p. 168-73.
86. Imai-Nishiya, H., et al., Double knockdown of alpha1,6-fucosyltransferase (FUT8) and GDP-mannose 4,6-dehydratase (GMD) in antibody-producing cells: a new strategy for generating fully non-fucosylated therapeutic antibodies with enhanced ADCC. BMC Biotechnol, 2007. 7: p. 84.
87. Kanda, Y., et al., Establishment of a GDP-mannose 4,6-dehydratase (GMD) knockout host cell line: a new strategy for generating completely non-fucosylated recombinant therapeutics. J Biotechnol, 2007. 130(3): p. 300-10.
88. Ripka, J., A. Adamany, and P. Stanley, Two Chinese hamster ovary glycosylation mutants affected in the conversion of GDP-mannose to GDP-fucose. Arch Biochem Biophys, 1986. 249(2): p. 533-45.
89. Kanda, Y., et al., Comparison of cell lines for stable production of fucose-negative antibodies with enhanced ADCC. Biotechnol Bioeng, 2006. 94(4): p. 680-8.
90. Narasimhan, S., Control of glycoprotein synthesis. UDP-GlcNAc:glycopeptide beta 4-N-acetylglucosaminyltransferase III, an enzyme in hen oviduct which adds GlcNAc in beta 1-4 linkage to the beta-linked mannose of the trimannosyl core of N-glycosyl oligosaccharides. J Biol Chem, 1982. 257(17): p. 10235-42.
91. Ferrara, C., et al., Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of Golgi enzyme localization domain and co-expression of heterologous beta1, 4-N-acetylglucosaminyltransferase III and Golgi alpha-mannosidase II. Biotechnol Bioeng, 2006. 93(5): p. 851-61.
92. Ferrara, C., et al., The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J Biol Chem, 2006. 281(8): p. 5032-6.
93. Davies, J., et al., Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FC gamma RIII. Biotechnol Bioeng, 2001. 74(4): p. 288-94.
94. Schuster, M., et al., Improved effector functions of a therapeutic monoclonal Lewis Y-specific antibody by glycoform engineering. Cancer Res, 2005. 65(17): p. 7934-41.
95. Umana, P., et al., Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol, 1999. 17(2): p. 176-80.
96. Kang, S., et al., Metabolic markers associated with high mannose glycan levels of therapeutic recombinant monoclonal antibodies. J Biotechnol, 2015. 203: p. 22-31.
97. Wright, A. and S.L. Morrison, Effect of altered CH2-associated carbohydrate structure on the functional properties and in vivo fate of chimeric mouse-human immunoglobulin G1. J Exp Med, 1994. 180(3): p. 1087-96.
98. Yu, M., et al., Production, characterization and pharmacokinetic properties of antibodies with N-linked Mannose-5 glycans. mAbs, 2012. 4(4): p. 475-487.
99. Reusch, D. and M.L. Tejada, Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology, 2015. 25(12): p. 1325-1334.
100. Zhong, X., et al., Engineering novel Lec1 glycosylation mutants in CHO–DUKX cells: Molecular insights and effector modulation of N-acetylglucosaminyltransferase I. Biotechnology and Bioengineering, 2012. 109(7): p. 1723-1734.
101. Chen, W. and P. Stanley, Five Lec1 CHO cell mutants have distinct Mgat1 gene mutations that encode truncated N-acetylglucosaminyltransferase I. Glycobiology, 2003. 13(1): p. 43-50.
102. Stanley, P., N-Acetylglucosaminyltransferase-I, in Handbook of Glycosyltransferases and Related Genes, N. Taniguchi, et al., Editors. 2002, Springer Japan. p. 61-69.
103. Stanley, P., Lectin-resistant CHO cells: selection of new mutant phenotypes. Somatic Cell Genet, 1983. 9(5): p. 593-608.
104. Stanley, P. and W. Chaney, Control of carbohydrate processing: the lec1A CHO mutation results in partial loss of N-acetylglucosaminyltransferase I activity. Mol Cell Biol, 1985. 5(6): p. 1204-11.
105. Wright, A., et al., In vivo trafficking and catabolism of IgG1 antibodies with Fc associated carbohydrates of differing structure. Glycobiology, 2000. 10(12): p. 1347-55.
106. Longmore, G.D. and H. Schachter, Product-identification and substrate-specificity studies of the GDP-L-fucose:2-acetamido-2-deoxy-beta-D-glucoside (FUC goes to Asn-linked GlcNAc) 6-alpha-L-fucosyltransferase in a Golgi-rich fraction from porcine liver. Carbohydr Res, 1982. 100: p. 365-92.
107. Voynow, J.A., et al., Purification and characterization of GDP-L-fucose-N-acetyl beta-D-glucosaminide alpha 1—-6fucosyltransferase from cultured human skin fibroblasts. Requirement of a specific biantennary oligosaccharide as substrate. Journal of Biological Chemistry, 1991. 266(32): p. 21572-21577.
108. Sealover, N.R., et al., Engineering Chinese hamster ovary (CHO) cells for producing recombinant proteins with simple glycoforms by zinc-finger nuclease (ZFN)-mediated gene knockout of mannosyl (alpha-1,3-)-glycoprotein beta-1,2-N-acetylglucosaminyltransferase (Mgat1). J Biotechnol, 2013. 167(1): p. 24-32.
109. Lee, J.S., et al., Site-specific integration in CHO cells mediated by CRISPR/Cas9 and homology-directed DNA repair pathway. Sci Rep, 2015. 5: p. 8572.
110. Elliott, S., et al., Control of rHuEPO biological activity: the role of carbohydrate. Exp Hematol, 2004. 32(12): p. 1146-55.
111. Meuris, L., et al., GlycoDelete engineering of mammalian cells simplifies N-glycosylation of recombinant proteins. Nat Biotech, 2014. 32(5): p. 485-489.
112. Lepenies, B. and P.H. Seeberger, Simply better glycoproteins. Nat Biotech, 2014. 32(5): p. 443-445.
113. Yang, Z., et al., Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat Biotech, 2015. 33(8): p. 842-844.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Pharmacology"
Pharmacology involves the study of drugs and how they affect the body. A pharmacologist contributes to drug development by researching and testing how the body reacts to medication, and whether the medication can have a positive impact on the body in terms of fighting illness and disease.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: