Synthesis of Octapeptin C4 and Analogues
Info: 8861 words (35 pages) Dissertation
Published: 9th Dec 2019
Tagged: ChemistryPharmacology
Summary:
Fungal infections have constantly increased over the last few decades, resulting in 1.5 million deaths worldwide per annum. Currently available antifungal drugs are no longer reliable due to poor efficacy, host toxicity and other regulatory restrictions. A class of cyclic lipopeptides called octapeptins have shown promising results against several microbial species. However, the complete antimicrobial spectrum of these compounds are yet to be discovered. In this regard, octapeptin C4, a subclass of the octapeptin family was synthesized along with its two analogues with new lipophilic groups: one from the echinocandin class of antifungal drug Micafungin, the other from a novel lipopeptide antibiotic, MX-2401. The yields of pure natural octapeptin C4 and Micafungin and MX-2401 analogues were 18.0%, 26.0% and 15.0% respectively. Followed by synthesis, in vitro assay of these compounds was performed against Cryptococcus neoformans.
1. Introduction:
1.1 Clinical Background:
Fungal infections have drastically increased since 1960s, affecting 1.2 billion people [1, 2]. These infections are prevalent worldwide, threatening lives of various immunocompromised patients (cancer, HIV/AIDS etc.) [3, 4]. Around 1.5 million people per annum die of invasive fungal infections; 90% of these are caused by the following fungal species: Aspergillus, Candida, Pneumocystis, Cryptococcus, Rhizopus and Mucor [4].
Fungal pathogens are often difficult to treat in mammalian hosts due to their similar eukaryotic cellular composition. Due to unavailability of fungal specific drug targets, there is always a high chance of host toxicity. Therefore, discovery of new, selective antifungal therapies is challenging [1, 4-6]. Options to treat fungal infections have been limited to the following: (1) membrane function disruptors (amphotericin B); (2) agents inhibiting DNA/RNA synthesis (flucytosine); (3) ergosterol synthesis inhibitors (azoles) and (4) glucan synthesis inhibitors (echinocandins) [7]. However, these current therapies are hindered by poor efficacy, side effects and toxicity [4, 8]. Therefore, it is necessary for scientists to search for alternative therapies to combat fungal infections.
Cryptococcosis is a type of systemic mycosis, caused by Cryptococcus neoformans [9]. C. neoformans is a pathogenic fungus responsible for deadly mycotic infections in AIDS patients, particularly in sub-Saharan Africa [10, 11]. These infections can further lead to encephalitis, meningitis or meningoencephalitis, which are difficult to treat and need proper medical attention [9, 12].
The polysaccharide capsule of C. neoformans is negatively charged [13], and has a potential role in maintaining its virulence [14, 15]. It guards the fungal cell against phagocytosis and facilitates opportunistic infections [15, 16]. In addition, the capsule enables the fungal cell to disseminate by both surviving and replicating in the lysosome [17]. Therefore, due to the capsule and a variety of other survival mechanisms [18, 19], cryptococcal infections can rapidly spread through the body and eventually reach the brain, causing cryptococcal meningitis [20].
According to WHO guidelines, the optimal treatment for cryptococcal meningitis begins with a two week course of amphotericin B along with flucytosine, followed by consolidation therapy with fluconazole [21]. However, flucytosine is expensive and unlicensed in many regions, like sub-Saharan Africa [21, 22]. In this case, therapy is confined to amphotericin B and fluconazole, or fluconazole monotherapy if amphotericin B is unavailable [22]. Unfortunately, a number of fluconazole resistant cases have been reported in recent years, which has made the fight against cryptococcal meningitis more urgent [22].
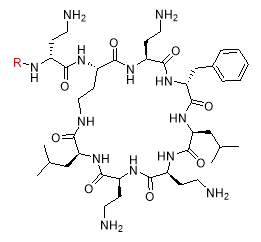
In 1970s, a class of cyclic lipopeptides called ‘octapeptins’ were discovered which showed efficacy against various microbial species, but little research has been made to explore its full antimicrobial spectrum [13]. Octapeptin C4 is a member of the octapeptin class, having a 3-hydroxy-decanoic acid moiety attached to the core structure [13, 23] (Fig. 2). It has been proposed that the negatively charged capsule of C. neoformans can concentrate cationic lipopeptides in the cell surface, thereby increasing fungal sensitivity to octapeptin C4 [13]. In addition, SAR of nine alanine scan derivatives of octapeptin C4 have revealed that the lipophilic groups as well as the cationic amino acid moieties are important for its antifungal properties [13]. To study the interaction between fungal capsule and octapeptin C4, it is desirable to synthesize analogues of these compounds, followed by in vitro assay against C. neoformans. The synthesis of these compounds can be achieved using a suitable technique like Solid Phase Peptide Synthesis (SPPS).
1.2 An overview of the Solid Phase Peptide Synthesis:
The importance of synthetic peptides in research and as therapeutics can hardly be denied. Synthesis of biologically active peptides provides opportunities to obtain novel compounds with improved biological properties and paves the way to study structure-activity relationships. Recombinant technologies are used to synthesize large peptides, whereas Solid Phase Peptide Synthesis (SPPS) is the most feasible approach for the synthesis of small peptides [24].
The major theme for SPPS is based on the elongation of a peptide chain in multiple steps, where the first amino acid is covalently linked to a solid support (resin) via a linker [25, 26] (Fig. 1). After the first amino acid is coupled to the resin, the α-amino protecting group is removed, followed by the addition of the second amino acid, resulting in the formation of a peptide bond [24, 25]. In this process, a series of amino acids are added to the growing peptide chain in a stepwise progression, involving a number of repeated deprotection and coupling reactions until the target peptide is assembled [25]. Finally, the peptide is cleaved off the resin, isolated and purified using suitable techniques [24, 25].
SPPS has got some major advantages over other conventional synthetic techniques [24, 25] as follows:






























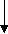




 Fig. 1. Diagrammatic representation of SPPS (Adapted from Merrifield, R. B. [27])
1.3 Fmoc SPPS vs. Boc SPPS:
In order to protect the amino acids, two strategies are widely used: (a) the butyloxycarbonyl (Boc)-protecting group or (b) 9-fluorenyl methoxy carbonyl (Fmoc) group [24]. The Boc protecting group is acid-labile and protects the α-amino group whereas the side chains are protected by orthogonal protecting groups such as benzylic groups. The Boc and benzylic groups can be cleaved by trifluoro acetic acid (TFA) and hydrogen fluoride (HF) respectively. On contrary, the Fmoc protecting group is base-labile, protecting the α-amino group, but the side chains are protected by acid labile tert-butyl or trityl-based groups. Fmoc can be cleaved by 20% piperidine in dimethylformamide (DMF), whereas removal of tert-butyl groups is achieved by TFA [24].
Fmoc-SPPS is preferred to Boc-SPPS, since the later involves use of HF which is hazardous, and the TFA used for Boc-removal may result in peptide loss at each synthetic step. Fmoc deprotection releases a fluorene group which has strong UV absorption properties. Therefore, Fmoc-SPPS is considered as a truly orthogonal protecting strategy for the production of smaller peptides [24, 28].
1.4 Objectives of the research project:
Previously, SPPS was applied to synthesize natural octapeptin C4 and other alanine scan derivatives [13]. Therefore, synthesis of octapeptin C4 and analogues will provide an opportunity to further characterize these compounds by structure activity relationship (SAR) studies.
This research project aims to investigate the following:
Fig. 1. Diagrammatic representation of SPPS (Adapted from Merrifield, R. B. [27])
1.3 Fmoc SPPS vs. Boc SPPS:
In order to protect the amino acids, two strategies are widely used: (a) the butyloxycarbonyl (Boc)-protecting group or (b) 9-fluorenyl methoxy carbonyl (Fmoc) group [24]. The Boc protecting group is acid-labile and protects the α-amino group whereas the side chains are protected by orthogonal protecting groups such as benzylic groups. The Boc and benzylic groups can be cleaved by trifluoro acetic acid (TFA) and hydrogen fluoride (HF) respectively. On contrary, the Fmoc protecting group is base-labile, protecting the α-amino group, but the side chains are protected by acid labile tert-butyl or trityl-based groups. Fmoc can be cleaved by 20% piperidine in dimethylformamide (DMF), whereas removal of tert-butyl groups is achieved by TFA [24].
Fmoc-SPPS is preferred to Boc-SPPS, since the later involves use of HF which is hazardous, and the TFA used for Boc-removal may result in peptide loss at each synthetic step. Fmoc deprotection releases a fluorene group which has strong UV absorption properties. Therefore, Fmoc-SPPS is considered as a truly orthogonal protecting strategy for the production of smaller peptides [24, 28].
1.4 Objectives of the research project:
Previously, SPPS was applied to synthesize natural octapeptin C4 and other alanine scan derivatives [13]. Therefore, synthesis of octapeptin C4 and analogues will provide an opportunity to further characterize these compounds by structure activity relationship (SAR) studies.
This research project aims to investigate the following:


 The resin (250 mg, 0.18 mmol) was swollen in THF (5 mL) for 30 mins and drained under vacuum. A solution of 4% hydrazine hydrate in anhydrous DMF (3.25 mL, 8 equiv.) was added to the resin and agitated for 1 hour. The solvent was drained, and washed with DMF (3 x 2 mL), methanol (3 x 2 mL) and DCM (3 x 2 mL). The resulting resin was dried under vacuum overnight [23].
2.1.3 Peptide cleavage from resin:
The resin (250 mg, 0.18 mmol) was swollen in THF (5 mL) for 30 mins and drained under vacuum. A solution of 4% hydrazine hydrate in anhydrous DMF (3.25 mL, 8 equiv.) was added to the resin and agitated for 1 hour. The solvent was drained, and washed with DMF (3 x 2 mL), methanol (3 x 2 mL) and DCM (3 x 2 mL). The resulting resin was dried under vacuum overnight [23].
2.1.3 Peptide cleavage from resin:
* Zorbax Eclipse XDB-Phenyl column, 0.1% TFA in H2O/Acetonitrile mobile phase
2.1.5 Global deprotection and purification:
* Zorbax Eclipse XDB-Phenyl column, 0.1% TFA in H2O/Acetonitrile mobile phase
As shown in Table 3, successful purification of each crude was achieved with Gilson HPLC. A Zorbax Eclipse XDB-Phenyl column with 0.1% TFA in H2O as buffer A and 0.1% TFA in acetonitrile as buffer B was used with a gradient as follows: 5% buffer B, 4 mins; 5% to 100% buffer B, 11 mins; 100% buffer B, 2 mins; 100% to 5% buffer B, 2 mins. The final yields for each pure compound are given in Table 3.
Table 3. LC-MS analysis of the final compounds
* Zorbax Eclipse XDB-Phenyl column, 0.1% TFA in H2O/Acetonitrile mobile phase
2.2 Determination of MIC of final compounds:
2.2.1 Preparation of YNB media:
0.85 gm of Yeast Nitrogen Base (YNB, without amino acids) media was weighed and dissolved in 445 mL of water, followed by autoclaving for 24 mins. After autoclaving, 50 mL of 20% glucose and 5 mL of 1M ammonium sulfate solution was added to make a final volume of 500 mL [30].
2.2.2 Preparation of inoculum:
5 ml of autoclaved water was poured into a falcon tube. Three single colonies of C. neoformans were picked from a culture plate using a wooden applicator stick and resuspended into 5 mL water in the falcon tube. The tube was subjected to vortex to ensure that colonies were resuspended completely into solution [30].
2.2.3 Counting cells using hemocytometer:
The hemocytometer and coverslip were cleaned using 70% ethanol. 10 µL of cell suspension was added into the edge of the coverslip so that the solution is properly diffused into the grid of hemocytometer. The cells were allowed to rest for 1 minute and observed under microscope to count Colony Forming Unit (CFU)/mL [30]. The CFU was found to be 1.95 x 106/mL.
2.2.4 Preparation of 96 well plate:
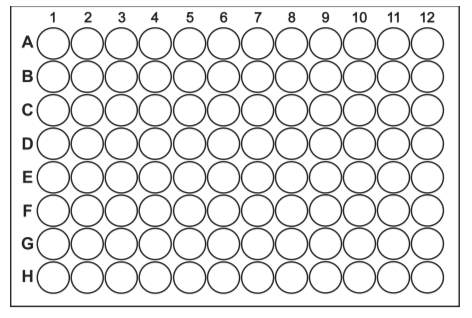 190 µL of YNB media was added to the column 1 (A-H) of the 96 well plate (Fig. 3). 100 µL of YNB media was added from column 2 to 12. A stock solution of 2 mg/mL was prepared for each of the following: Fluconazole and natural Octapeptin C4 as “control” and Micafungin analogue (R2) and MX-2401 analogue (R3) as “tests”. Fluconazole was provided by the courtesy of James Fraser’s Lab. 10 µL of the stock solution was added to the column 1 of each well (A-F) respectively (Fig. 3). The solutions were mixed well by pipetting up and down several times by using a multi-channel pipette. 100 µL of solution was transferred from column 1 to 2, mixed well by pipetting up and down several times, followed by adding 100 µL of solution from column 2 to 3 and continued up to column 11. In this way, serial dilution was performed from column 1 to 11 (Fig. 3). However, 100 µL solution from column 11 was discarded before adding to column 12, since it was used as negative control [30].
190 µL of YNB media was added to the column 1 (A-H) of the 96 well plate (Fig. 3). 100 µL of YNB media was added from column 2 to 12. A stock solution of 2 mg/mL was prepared for each of the following: Fluconazole and natural Octapeptin C4 as “control” and Micafungin analogue (R2) and MX-2401 analogue (R3) as “tests”. Fluconazole was provided by the courtesy of James Fraser’s Lab. 10 µL of the stock solution was added to the column 1 of each well (A-F) respectively (Fig. 3). The solutions were mixed well by pipetting up and down several times by using a multi-channel pipette. 100 µL of solution was transferred from column 1 to 2, mixed well by pipetting up and down several times, followed by adding 100 µL of solution from column 2 to 3 and continued up to column 11. In this way, serial dilution was performed from column 1 to 11 (Fig. 3). However, 100 µL solution from column 11 was discarded before adding to column 12, since it was used as negative control [30].
 In the mass spectrum of octapeptin C4 analogue with Micafungin tail (R2), it was evident that the most intense peak corresponded to MH+ (1608.0) and m/z 1508.0 corresponded to MH+-Boc (Fig. 4b).
In the mass spectrum of octapeptin C4 analogue with Micafungin tail (R2), it was evident that the most intense peak corresponded to MH+ (1608.0) and m/z 1508.0 corresponded to MH+-Boc (Fig. 4b).
 In the mass spectrum of octapeptin C4 analogue with MX-2401 tail (R3), it was evident that the most intense peak corresponded to MH+ (1562.0) and m/z 1462.0 corresponded to MH+-Boc (Fig. 4c).
In the mass spectrum of octapeptin C4 analogue with MX-2401 tail (R3), it was evident that the most intense peak corresponded to MH+ (1562.0) and m/z 1462.0 corresponded to MH+-Boc (Fig. 4c).
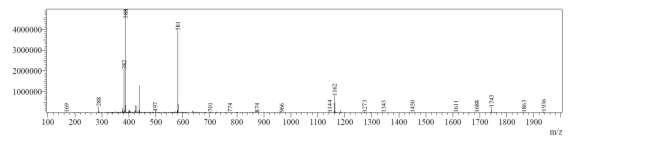



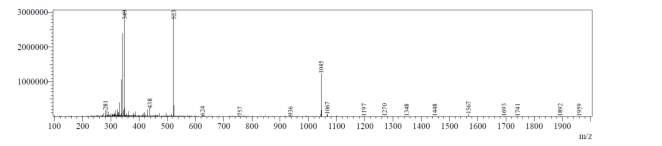
Appendix 1. HRMS exact mass (ESI microTOF-LC) of natural octapeptin C4 (R1)
 Appendix 2. HRMS exact mass (ESI microTOF-LC) of octapeptin C4 analogue (R2)
Appendix 2. HRMS exact mass (ESI microTOF-LC) of octapeptin C4 analogue (R2)
 Appendix 3. HRMS exact mass (ESI microTOF-LC) of octapeptin C4 analogue (R3)
Appendix 3. HRMS exact mass (ESI microTOF-LC) of octapeptin C4 analogue (R3)
 References:
1. Vandeputte, P., Ferrari, S., Coste, A. T., Antifungal Resistance and New Strategies to Control Fungal Infections. Int J Microbiol, 2012. 3: p. 1-26.
2. Chang, Y.L., Yu, S. J., Heitman, J., Wellington, M., Chen, Y. L., New Facets of Antifungal Therapy. Virulence, 2017. 8(2): p. 222-236.
3. May, R.C., Stone, N. R. H., Wiesner, D. L., Bicanic, T., Nielsen, K., Cryptococcus: From Environmental Saprophyte to Global Pathogen. Nat Rev Microbiol, 2016. 14(2): p. 106-117.
4. Campoy, S., Adrio, J. L., Antifungals. Biochem Pharmacol, 2017. 133: p. 86-96.
5. Scorzoni, L., de Paula e Silva, A. C. A., Marcos, C. M., Assato, P. A., de Melo, W. C. M. A., de Oliviera, H. C., Costa-Orlandi, C. B., Mendes-Giannini, M. J. S., Fusco-Almeida, A. M., Antifungal Therapy: New Advances in the Understanding and Treatment of Mycosis. Front Microbiol, 2017. 8(36): p. 1-23.
6. Roemer, T., Krysan, D. J., Antifungal Drug Development: Challenges, Unmet Clinical Needs and New Approaches. Cold Spring Harb Perspect Med, 2014. 4(5): p. 1-15.
7. Perea, S., Patterson, T. F., Antifungal Resistance in Pathogenic Fungi. Clin Infect Dis, 2002. 35(9): p. 1073-1080.
8. Kathiravan, M.K., Salake, A. B., Chothe, A. S., Dudhe, P. B., Watode, R. P., Mukta, M. S., Gadhwe, S., The Biology and Chemistry of Antifungal Agents: A review. Bioorg Med Chem, 2012. 20(19): p. 5678-5698.
9. Gullo, F.P., Rossi, S. A., Sardi, J. de C. O., Teodoro, V. L. I., Mendes-Giannini, M. J. S., Fusco-Almeida, A. M., Cryptococcosis: Epidemiology, Fungal Resistance and New Alternatives for Treatment. Eur J Clin Microbiol Infect Dis, 2013. 32(11): p. 1377-1391.
10. Archibald, L.K., Tuohy, M. J., Wilson, D. A., Nwanyanwu, O., Kazembe, P. N., Tansuphasawadikul, S., Eampokalap, B., Chaovavanich, A., Reller, L. B., Jarvis, W. R., Hall, G. S., Procop, G. W., Antifungal Susceptibilities of Cryptococcus neoformans. Emerg Infect Dis, 2004. 10(1): p. 143-145.
11. Rajasingham, R., Smith, R. M., Park, B. J., Jarvis, J. N., Govender, N. P., Chiller, T. M., Denning, D. W., Loyse, A., Boulware, D. R., Global Burden of Disease of HIV-associated Cryptococcal Meningitis: An Updated Analysis. Lancet Infect Dis, 2017. 17: p. 873-881.
12. Mourad, A., Perfect, J. R., Present and Future Therapy of Cryptococcus Infections. J Fungi, 2018. 4(79): p. 1-10.
13. Chitty, J.L., Butler, M. S., Suboh, A., Edwards, D. J., Cooper, M. A., Fraser, J. A., Robertson, A. A. B., Antimicrobial Octapeptin C4 Analogues Active Against Cryptococcus Species. Antimicrob Agents Chemother, 2018. 62(2): p. 1-10.
14. Zaragoza, O., Telzak, A., Bryan, R. A., Dadachova, E., Casadevall, A., The Polysaccharide Capsule of the Pathogenic Fungus Cryptococcus neoformans Enlarges by Distal Growth and is Rearranged During Budding. Mol Microbiol, 2006. 59(1): p. 67-83.
15. Bose, I., Reese, A. J., Ory, J. J., Janbon, G., Doering, T. L., A Yeast under Cover: the Capsule of Cryptococcus neoformans. Eukaryot Cell, 2003. 2(4): p. 655-663.
16. Cherniak, R., Sundstrom, J. B., Polysaccharide Antigens of the Capsule of Cryptococcus neoformans. Infect Immun, 1994. 62(5): p. 1507-1512.
17. Davis, M.J., Eastman, A. J., Qiu, Y., Gregorka, B., Kozel, T. R., Osterholzer, J. J., Curtis, J. L., Swanson, J. A., Olszweski, M. A., Cryptococcus neoformans–Induced Macrophage Lysosome Damage Crucially Contributes to Fungal Virulence. J Immunol, 2015. 194(5): p. 2219-2231.
18. Kronstad, J., Saikia, S., Nielson, E. D., Kretschmer, M., Jung, W., Hu, G., Geddes, J. M. H., Griffiths, E. J., Choi, J., Cadiuex, B., Caza, M., Attarian, R., Adaptation of Cryptococcus neoformans to Mammalian Hosts: Integrated Regulation of Metabolism and Virulence. Eukaryot Cell, 2012. 11(2): p. 109-118.
19. Alanio, A., Vernel-Pauillac, F., Sturny-Leclere, A., Dromer, F., Cryptococcus neoformans Host Adaptation: Toward Biological Evidence of Dormancy. mBio, 2015. 6(2): p. 1-13.
20. Yang, C.L., Wang, J., Zou, L. L., Innate Immune Evasion Strategies Against Cryptococcal Meningitis Caused by Cryptococcus neoformans. Exp Ther Med, 2017. 14(6): p. 5243-5250.
21. Smith, K.D., Achan, B., Hullsiek, K. H., McDonald, T. R., Okagaki, L. H., Alhadab, A. A., Akampurira, A., Rhein, J. R., Meya, D. B., Boulware, D. R., Nielsen, K., Increased Antifungal Drug Resistance in Clinical Isolates of Cryptococcus neoformans in Uganda. Antimicrob Agents Chemother, 2015. 59: p. 7197-7204.
22. Mpoza, E., Rhein, J., Abassi, M., Emerging Fluconazole Resistance: Implications for the Management of Cryptococcal Meningitis. Med Mycol Case Rep, 2018. 19: p. 30-32.
23. Becker, B., Butler, M. S., Hansford, K. A., Gallardo-Godoy, A., Elliott, A. G., Huang, J. X., Edwards, D. J., Blaskovich, M. A. T., Cooper, M. A., Synthesis of Octapeptin C4 and Biological Profiling Aganist NDM-1 and Polymyxin Resistant Bacteria. Bioorg Med Chem Lett, 2017. 27(11): p. 2407-2409.
24. Hansen, P.R., Oddo, A., Fmoc Solid-Phase Peptide Synthesis, in Peptide Antibodies-Methods and Protocols, G. Houen, Editor. 2015, Humana Press: Statens Serum Institut, Copenhagen, Denmark. p. 33-50.
25. Merrifield, R.B., Solid Phase Peptide Synthesis, in Advances in Enzymology and Related Areas of Molecular Biology, F.F. Nord, Editor. 1969, John Wiley & Sons, Inc.: New York City. p. 221-295.
26. Fields, G.B., Noble, R. L., Solid Phase Peptide Synthesis utilizing 9-fluorenylmethoxycarbonyl Amino Acids. Int J Peptide Protein Res, 1990. 35: p. 161-214.
27. Merrifield, R.B., Solid Phase Peptide Synthesis, in The Chemistry of Polypeptides, P.G. Katsoyannis, Editor. 1973, Plenum Press: New York-London. p. 335-362.
28. Behrendt, R., White, P., Offer, J., Advances in Fmoc Solid-Phase Peptide Synthesis. J Pept Sci, 2016. 22: p. 4-27.
29. Merck. New Orthogonally Protected Lysine Derivatives. 2018 [cited 2018 16 September]; Available from: www.merckmillipore.com/novabiochem.
30. Fraser, J., MIC Laboratory Protocol. 2018, The University of Queenland.
31. ThermoFisher. Liquid Chromatography Mass Spectrometry (LC-MS) Information. 2018 [cited 2018 14 September]; Available from: https://www.thermofisher.com/au/en/home/industrial/mass-spectrometry/mass-spectrometry-learning-center/liquid-chromatography-mass-spectrometry-lc-ms-information.html.
32. Chemyx. An overview of HPLC, MS, and LC-MS. 2018 [cited 2018 14 September]; Available from: https://www.chemyx.com/support/knowledge-base/applications/basic-principles-hplc-ms-lc-ms/.
33. Guy, C.A., Fields, G. B., Trifluoroacetic Acid Cleavage and Deprotection of Resin-bound Peptides Following Synthesis by Fmoc Chemistry. Methods Enzymol, 1997. 289: p. 67-83.
34. Pearson, D.A., Blanchette, M., Baker, M. L., Guindon, C. A., Trialkylsilanes as Scavengers For The Trifluoroacetic acid Deblocking of Protecting Groups in Peptide Synthesis. Tetrahedron Lett, 1989. 30(21): p. 2739-2742.
35. Pleil, J.D., Isaacs, K. K., High Resolution Mass Spectrometry: Basic Principles for Using Exact Mass and Mass Defect for Discovery Analysis of Organic Molecules in Blood, Breath, Urine and Environmental Media. J Breath Res, 2016. 10: p. 1-10.
36. Stock, N.L., Introducing Graduate Students to High-Resolution Mass Spectrometry (HRMS) Using a Hands-On Approach. J Chem Educ, 2017. 94: p. 1978-1982.
References:
1. Vandeputte, P., Ferrari, S., Coste, A. T., Antifungal Resistance and New Strategies to Control Fungal Infections. Int J Microbiol, 2012. 3: p. 1-26.
2. Chang, Y.L., Yu, S. J., Heitman, J., Wellington, M., Chen, Y. L., New Facets of Antifungal Therapy. Virulence, 2017. 8(2): p. 222-236.
3. May, R.C., Stone, N. R. H., Wiesner, D. L., Bicanic, T., Nielsen, K., Cryptococcus: From Environmental Saprophyte to Global Pathogen. Nat Rev Microbiol, 2016. 14(2): p. 106-117.
4. Campoy, S., Adrio, J. L., Antifungals. Biochem Pharmacol, 2017. 133: p. 86-96.
5. Scorzoni, L., de Paula e Silva, A. C. A., Marcos, C. M., Assato, P. A., de Melo, W. C. M. A., de Oliviera, H. C., Costa-Orlandi, C. B., Mendes-Giannini, M. J. S., Fusco-Almeida, A. M., Antifungal Therapy: New Advances in the Understanding and Treatment of Mycosis. Front Microbiol, 2017. 8(36): p. 1-23.
6. Roemer, T., Krysan, D. J., Antifungal Drug Development: Challenges, Unmet Clinical Needs and New Approaches. Cold Spring Harb Perspect Med, 2014. 4(5): p. 1-15.
7. Perea, S., Patterson, T. F., Antifungal Resistance in Pathogenic Fungi. Clin Infect Dis, 2002. 35(9): p. 1073-1080.
8. Kathiravan, M.K., Salake, A. B., Chothe, A. S., Dudhe, P. B., Watode, R. P., Mukta, M. S., Gadhwe, S., The Biology and Chemistry of Antifungal Agents: A review. Bioorg Med Chem, 2012. 20(19): p. 5678-5698.
9. Gullo, F.P., Rossi, S. A., Sardi, J. de C. O., Teodoro, V. L. I., Mendes-Giannini, M. J. S., Fusco-Almeida, A. M., Cryptococcosis: Epidemiology, Fungal Resistance and New Alternatives for Treatment. Eur J Clin Microbiol Infect Dis, 2013. 32(11): p. 1377-1391.
10. Archibald, L.K., Tuohy, M. J., Wilson, D. A., Nwanyanwu, O., Kazembe, P. N., Tansuphasawadikul, S., Eampokalap, B., Chaovavanich, A., Reller, L. B., Jarvis, W. R., Hall, G. S., Procop, G. W., Antifungal Susceptibilities of Cryptococcus neoformans. Emerg Infect Dis, 2004. 10(1): p. 143-145.
11. Rajasingham, R., Smith, R. M., Park, B. J., Jarvis, J. N., Govender, N. P., Chiller, T. M., Denning, D. W., Loyse, A., Boulware, D. R., Global Burden of Disease of HIV-associated Cryptococcal Meningitis: An Updated Analysis. Lancet Infect Dis, 2017. 17: p. 873-881.
12. Mourad, A., Perfect, J. R., Present and Future Therapy of Cryptococcus Infections. J Fungi, 2018. 4(79): p. 1-10.
13. Chitty, J.L., Butler, M. S., Suboh, A., Edwards, D. J., Cooper, M. A., Fraser, J. A., Robertson, A. A. B., Antimicrobial Octapeptin C4 Analogues Active Against Cryptococcus Species. Antimicrob Agents Chemother, 2018. 62(2): p. 1-10.
14. Zaragoza, O., Telzak, A., Bryan, R. A., Dadachova, E., Casadevall, A., The Polysaccharide Capsule of the Pathogenic Fungus Cryptococcus neoformans Enlarges by Distal Growth and is Rearranged During Budding. Mol Microbiol, 2006. 59(1): p. 67-83.
15. Bose, I., Reese, A. J., Ory, J. J., Janbon, G., Doering, T. L., A Yeast under Cover: the Capsule of Cryptococcus neoformans. Eukaryot Cell, 2003. 2(4): p. 655-663.
16. Cherniak, R., Sundstrom, J. B., Polysaccharide Antigens of the Capsule of Cryptococcus neoformans. Infect Immun, 1994. 62(5): p. 1507-1512.
17. Davis, M.J., Eastman, A. J., Qiu, Y., Gregorka, B., Kozel, T. R., Osterholzer, J. J., Curtis, J. L., Swanson, J. A., Olszweski, M. A., Cryptococcus neoformans–Induced Macrophage Lysosome Damage Crucially Contributes to Fungal Virulence. J Immunol, 2015. 194(5): p. 2219-2231.
18. Kronstad, J., Saikia, S., Nielson, E. D., Kretschmer, M., Jung, W., Hu, G., Geddes, J. M. H., Griffiths, E. J., Choi, J., Cadiuex, B., Caza, M., Attarian, R., Adaptation of Cryptococcus neoformans to Mammalian Hosts: Integrated Regulation of Metabolism and Virulence. Eukaryot Cell, 2012. 11(2): p. 109-118.
19. Alanio, A., Vernel-Pauillac, F., Sturny-Leclere, A., Dromer, F., Cryptococcus neoformans Host Adaptation: Toward Biological Evidence of Dormancy. mBio, 2015. 6(2): p. 1-13.
20. Yang, C.L., Wang, J., Zou, L. L., Innate Immune Evasion Strategies Against Cryptococcal Meningitis Caused by Cryptococcus neoformans. Exp Ther Med, 2017. 14(6): p. 5243-5250.
21. Smith, K.D., Achan, B., Hullsiek, K. H., McDonald, T. R., Okagaki, L. H., Alhadab, A. A., Akampurira, A., Rhein, J. R., Meya, D. B., Boulware, D. R., Nielsen, K., Increased Antifungal Drug Resistance in Clinical Isolates of Cryptococcus neoformans in Uganda. Antimicrob Agents Chemother, 2015. 59: p. 7197-7204.
22. Mpoza, E., Rhein, J., Abassi, M., Emerging Fluconazole Resistance: Implications for the Management of Cryptococcal Meningitis. Med Mycol Case Rep, 2018. 19: p. 30-32.
23. Becker, B., Butler, M. S., Hansford, K. A., Gallardo-Godoy, A., Elliott, A. G., Huang, J. X., Edwards, D. J., Blaskovich, M. A. T., Cooper, M. A., Synthesis of Octapeptin C4 and Biological Profiling Aganist NDM-1 and Polymyxin Resistant Bacteria. Bioorg Med Chem Lett, 2017. 27(11): p. 2407-2409.
24. Hansen, P.R., Oddo, A., Fmoc Solid-Phase Peptide Synthesis, in Peptide Antibodies-Methods and Protocols, G. Houen, Editor. 2015, Humana Press: Statens Serum Institut, Copenhagen, Denmark. p. 33-50.
25. Merrifield, R.B., Solid Phase Peptide Synthesis, in Advances in Enzymology and Related Areas of Molecular Biology, F.F. Nord, Editor. 1969, John Wiley & Sons, Inc.: New York City. p. 221-295.
26. Fields, G.B., Noble, R. L., Solid Phase Peptide Synthesis utilizing 9-fluorenylmethoxycarbonyl Amino Acids. Int J Peptide Protein Res, 1990. 35: p. 161-214.
27. Merrifield, R.B., Solid Phase Peptide Synthesis, in The Chemistry of Polypeptides, P.G. Katsoyannis, Editor. 1973, Plenum Press: New York-London. p. 335-362.
28. Behrendt, R., White, P., Offer, J., Advances in Fmoc Solid-Phase Peptide Synthesis. J Pept Sci, 2016. 22: p. 4-27.
29. Merck. New Orthogonally Protected Lysine Derivatives. 2018 [cited 2018 16 September]; Available from: www.merckmillipore.com/novabiochem.
30. Fraser, J., MIC Laboratory Protocol. 2018, The University of Queenland.
31. ThermoFisher. Liquid Chromatography Mass Spectrometry (LC-MS) Information. 2018 [cited 2018 14 September]; Available from: https://www.thermofisher.com/au/en/home/industrial/mass-spectrometry/mass-spectrometry-learning-center/liquid-chromatography-mass-spectrometry-lc-ms-information.html.
32. Chemyx. An overview of HPLC, MS, and LC-MS. 2018 [cited 2018 14 September]; Available from: https://www.chemyx.com/support/knowledge-base/applications/basic-principles-hplc-ms-lc-ms/.
33. Guy, C.A., Fields, G. B., Trifluoroacetic Acid Cleavage and Deprotection of Resin-bound Peptides Following Synthesis by Fmoc Chemistry. Methods Enzymol, 1997. 289: p. 67-83.
34. Pearson, D.A., Blanchette, M., Baker, M. L., Guindon, C. A., Trialkylsilanes as Scavengers For The Trifluoroacetic acid Deblocking of Protecting Groups in Peptide Synthesis. Tetrahedron Lett, 1989. 30(21): p. 2739-2742.
35. Pleil, J.D., Isaacs, K. K., High Resolution Mass Spectrometry: Basic Principles for Using Exact Mass and Mass Defect for Discovery Analysis of Organic Molecules in Blood, Breath, Urine and Environmental Media. J Breath Res, 2016. 10: p. 1-10.
36. Stock, N.L., Introducing Graduate Students to High-Resolution Mass Spectrometry (HRMS) Using a Hands-On Approach. J Chem Educ, 2017. 94: p. 1978-1982.
- A simplified and accelerated multistep synthesis, providing the reaction to be completed in a single reaction vessel
- The use of excess amino acids drives the reaction to full completion with minimal chance of product loss, thereby minimizing the need for purification of each intermediate product
- High yield of the final product since excess reagents are used to ensure full completion of each reaction step
Solid support (Resin)
Amino acids
Protecting groups
Cleavage from resin, isolation, purification
Fig. 1. Diagrammatic representation of SPPS (Adapted from Merrifield, R. B. [27])
1.3 Fmoc SPPS vs. Boc SPPS:
In order to protect the amino acids, two strategies are widely used: (a) the butyloxycarbonyl (Boc)-protecting group or (b) 9-fluorenyl methoxy carbonyl (Fmoc) group [24]. The Boc protecting group is acid-labile and protects the α-amino group whereas the side chains are protected by orthogonal protecting groups such as benzylic groups. The Boc and benzylic groups can be cleaved by trifluoro acetic acid (TFA) and hydrogen fluoride (HF) respectively. On contrary, the Fmoc protecting group is base-labile, protecting the α-amino group, but the side chains are protected by acid labile tert-butyl or trityl-based groups. Fmoc can be cleaved by 20% piperidine in dimethylformamide (DMF), whereas removal of tert-butyl groups is achieved by TFA [24].
Fmoc-SPPS is preferred to Boc-SPPS, since the later involves use of HF which is hazardous, and the TFA used for Boc-removal may result in peptide loss at each synthetic step. Fmoc deprotection releases a fluorene group which has strong UV absorption properties. Therefore, Fmoc-SPPS is considered as a truly orthogonal protecting strategy for the production of smaller peptides [24, 28].
1.4 Objectives of the research project:
Previously, SPPS was applied to synthesize natural octapeptin C4 and other alanine scan derivatives [13]. Therefore, synthesis of octapeptin C4 and analogues will provide an opportunity to further characterize these compounds by structure activity relationship (SAR) studies.
This research project aims to investigate the following:
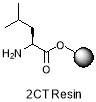
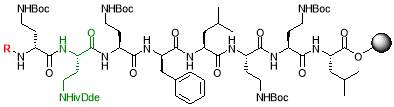
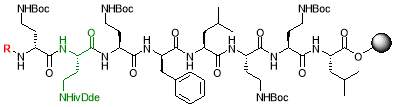
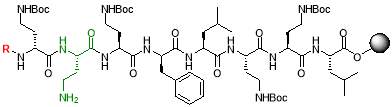
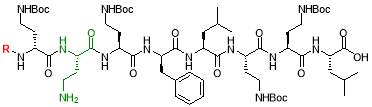
- Synthesis of octapeptin C4 and analogues with novel lipid groups using “Fmoc Solid Phase Peptide Synthesis” (Fmoc-SPPS). Two novel lipid groups were used to synthesize octapeptin C4 analogues: one from the echinocandin class of antifungal drug Micafungin, the other from a novel lipopeptide antibiotic, MX-2401 (Fig. 2).
- In vitro assay of octapeptin C4 and analogues against C. neoformans.
Fig. 2. Different octapeptin C4 analogues
2. Materials and Methods:
2.1 Synthetic route to octapeptin C4:
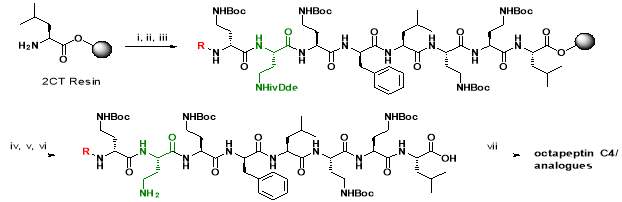
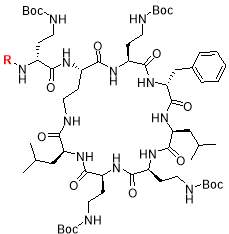
Scheme 1: Solid phase synthesis and off-resin cyclisation of octapeptin C4. Reagents: i) amino acids, HBTU, DIPEA, DMF; ii) 20% piperidine in DMF; iii) Lipophilic moiety (R1,2,3), HBTU, DIPEA, DMF; iv) 4% hydrazine hydrate in DMF; v) 20% HFIP in DCM; vi) DPPA, DIPEA, DMF; vii) TFA/H2O/TIS
Octapeptin C4 and analogues were synthesized by Fmoc-SPPS, in a route developed by Becker et al. [23]. The peptide was synthesized on a 2-chlorotrityl (2-CT) resin, using Fmoc-protected amino acids on the N-terminal. The amide side chains on 2, 4-diaminobutyric acid (Dab) were protected by a Boc group. Orthogonal ivDde protection on Dab (marked green in Scheme 1) allowed the amino group to be selectively deprotected without affecting other Boc-protected groups [23]. A unique feature of ivDde is that, it is stable to 20% piperidine in DMF but cleaved by 4% hydrazine hydrate in DMF [29]. This enables the amino groups of ivDde-protected residues to be selectively deprotected without affecting side chain protecting groups on the other residues. In addition, the deprotection reaction creates a UV-active indazole by-product, which can be easily monitored by spectrophotometry [29].
Followed by orthogonal ivDde protection, the resin was divided and lipid moieties (R1,2,3, Fig. 2) were added. The ivDde protecting group was cleaved by 4% hydrazine, followed by resin cleavage with a solution of 20% hexafluoroisopropanol (HFIP) in dichloromethane (DCM). Cyclization of the peptide was achieved using diphenyl phosphoryl azide (DPPA), diisopropylethylamine (DIPEA) base in dimethylformamide (DMF). Global deprotection using trifluoroacetic acid (TFA) and purification by HPLC yielded the final octapeptin C4 analogues [23].
2.1.1 Fmoc Solid phase peptide synthesis (Fmoc SPPS):
i) Amino acids, HBTU, DIPEA, DMF
ii) 20% piperidine in DMF
iii) Lipophilic moiety (R1,2,3),HBTU, DIPEA, DMF
The 2-CT resin with L-leu (1.5 gm, 1.08 mmol, loading = 0.72 mmol/g) was suspended in DMF (4 mL). The Fmoc-protected amino acid (1 equiv.) was dissolved in DMF (2 mL) and treated with DIPEA (356 µL, 4 equiv.) and a solution of HBTU in 0.5M DMF (2.16 mL, 2 equiv.). The solution was agitated for 1 min and added to the suspended resin. The reaction was shaken for 2 hours, drained, washed with DMF (3 x 2 mL), methanol (3 x 2 mL) and DCM (3 x 2 mL) and left to dry in vacuo for 15 mins. The entire procedure was repeated with an additional equivalent of amino acid to ensure complete coupling. Finally, the resin was treated with 20% piperidine in DMF (4 mL) and agitated for 30 mins to remove the Fmoc protecting group, so that the next amino acid can be added to the growing peptide chain. The resin was drained, and washed as before with DMF, methanol and DCM [23]. This procedure was repeated to couple each amino acid. After completion of peptide synthesis, the resin was divided and all lipid groups (R1,2,3) were added (1.5 equiv.) using the similar protocol as mentioned.
In order to check the coupling efficiency at each synthetic step, 10 µl of 95: 5: 5 TFA/H2O/TIS was added to a small amount of resin (~5 mg) and kept for 10 mins to clear the peptide from the resin, followed by addition of 50: 50 acetonitrile/H2O (1 mL), filtered and finally LC-MS was performed [23].
2.1.2 Deprotection of ivDde from resin:
4% hydrazine hydrate in DMF
The resin (250 mg, 0.18 mmol) was swollen in THF (5 mL) for 30 mins and drained under vacuum. A solution of 4% hydrazine hydrate in anhydrous DMF (3.25 mL, 8 equiv.) was added to the resin and agitated for 1 hour. The solvent was drained, and washed with DMF (3 x 2 mL), methanol (3 x 2 mL) and DCM (3 x 2 mL). The resulting resin was dried under vacuum overnight [23].
2.1.3 Peptide cleavage from resin:
20% HFIP in DCM
The resin (250 mg, 0.18 mmol) was treated with a cleavage solution, comprising of 20% HFIP in DCM (1:4, 20 mL) for 1 hour. The resulting suspension was drained and resin was shaken with an additional volume of cleavage solution (10 mL) for 30 mins. The solvent was drained and washed with DCM (3 x 3 mL). The filtrates were combined and concentrated in vacuo to afford the crude product [23].
2.1.4 Cyclization of the peptide:
DPPA, DIPEA, DMF
The cleaved peptide was treated with DMF (8 mL), DPPA (4 equiv.) and DIPEA (8 equiv.) and stirred at 60ºC overnight for cyclization. All compounds were purified directly from reaction mixture using GRACE® Reveleris medium pressure liquid chromatography (MPLC) system with C-18 cartridge and acetonitrile/H2O as mobile phase [23]. After LC-MS, all appropriate fractions were combined and freeze dried to give final products (Table 1).
Table 1. LC-MS analysis of the cyclized peptide
| Compound with lipid group | Amount (mg) | Retention time* (tR, min) | Yield (%) | Purity by UV (%) | (+)(ES) m/z | |
| MH+ | MH+-Boc | |||||
| R1 | 36.0 | 3.507 | 19.0 | 100.0 | 1445.0 | 1345.0 |
| R2 | 132.0 | 5.482 | 30.0 | 80.0 | 1608.0 | 1508.0 |
| R3 | 116.0 | 4.045 | 28.0 | 80.0 | 1562.0 | 1462.0 |
TFA/H2O/TIS
Each cyclized peptide was treated with 90%TFA/5%H2O/5%TIS (3 mL), stirred for 30 mins, then concentrated in vacuo to obtain the crude product [23]. The resulting mass of each crude is presented in Table 2.
Table 2. LC-MS analysis of the crude deprotected peptides
| Compound with lipid group | Amount (mg) | Retention time* (tR, min) | (+)(ES) m/z | ||
| MH+ | MH22+ | MH33+ | |||
| R1 | 95.0 | 2.920 | 1045.0 | 523.0 | 349.0 |
| R2 | 222.0 | 2.889 | 1208.0 | 604.0 | 403.3 |
| R3 | 193.0 | 2.849 | 1162.0 | 581.0 | 387.9 |
| Compound with lipid group | Amount (mg) | Retention time* (tR, min) | Yield (%) | Purity by UV (%) |
| R1 | 33.0 | 2.626 | 18.0 | 100.0 |
| R2 | 113.0 | 2.923 | 26.0 | 100.0 |
| R3 | 62.0 | 2.862 | 15.0 | 100.0 |
190 µL of YNB media was added to the column 1 (A-H) of the 96 well plate (Fig. 3). 100 µL of YNB media was added from column 2 to 12. A stock solution of 2 mg/mL was prepared for each of the following: Fluconazole and natural Octapeptin C4 as “control” and Micafungin analogue (R2) and MX-2401 analogue (R3) as “tests”. Fluconazole was provided by the courtesy of James Fraser’s Lab. 10 µL of the stock solution was added to the column 1 of each well (A-F) respectively (Fig. 3). The solutions were mixed well by pipetting up and down several times by using a multi-channel pipette. 100 µL of solution was transferred from column 1 to 2, mixed well by pipetting up and down several times, followed by adding 100 µL of solution from column 2 to 3 and continued up to column 11. In this way, serial dilution was performed from column 1 to 11 (Fig. 3). However, 100 µL solution from column 11 was discarded before adding to column 12, since it was used as negative control [30].
A= Fluconazole
B= Octapeptin C4 (R1)
C= Micafungin (R2) (Pure)*
D= MX-2401 (R3) (Pure)*
E= Micafungin (R2) (Trace)*
F= MX-2401 (R3) (Trace)*
G= (None)
H= (None)
* Based on HPLC purification
Fig. 3. Diagrammatic representation of 96 well plate for MIC assay of final compounds
Followed by serial dilution, the inoculum was diluted 1000 times (10 µL of inoculum into 10 mL of YNB media). 100 µL of the inoculum was added into each well from column 1 to 12. Therefore, the final volume in each well from column 1 to 12 was 200 µL, with highest concentration of the compound in column 1 (100 µg/mL) to the lowest in column 11 (0.098 µg/mL). The 96-well plate was placed in incubator at 35ºC and growth of C. neoformans was observed after 48 hours [30].
3. Results and Discussion:
3.1 Characterization of the protected cyclized peptide:
Liquid Chromatography (LC) is a useful technique for separation of sample according to their physico-chemical properties. Hence it is often combined with ionization in Mass spectroscopy (MS) to ensure effective separation of required compounds with minimal experimental error and improved accuracy [31, 32].
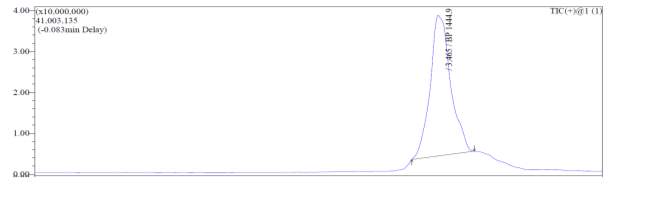
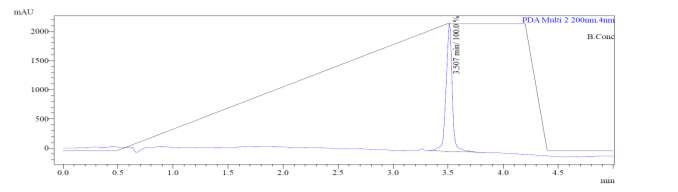
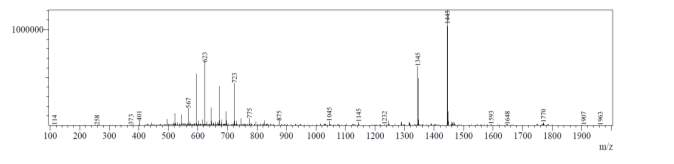
After first-pass GRACE® MPLC purification, each of the cyclized peptides were characterized by LC-MS. In the mass spectrum of natural octapeptin C4 (R1), it was evident that the most intense peak corresponded to MH+ (1445.0) and m/z 1345.0 corresponded to MH+-Boc (Fig. 4a).
A
B
C
MH+ (1445)
D
MH+-Boc (1345)
Fig. 4a. LC-MS of cyclized natural octapeptin C4 (R1) A.ELSD B. TIC C. UV (200 nm) D. (+)-ESI-MS
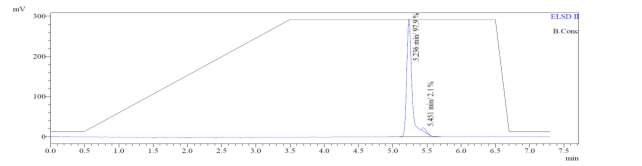
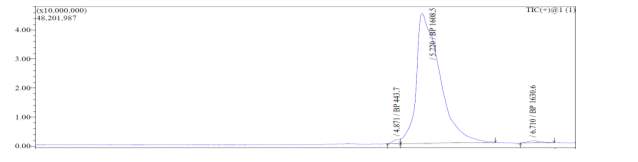
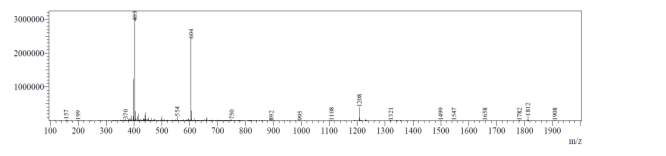
In the mass spectrum of octapeptin C4 analogue with Micafungin tail (R2), it was evident that the most intense peak corresponded to MH+ (1608.0) and m/z 1508.0 corresponded to MH+-Boc (Fig. 4b).
A
B
C
MH+ (1608)
MH+-Boc (1508)
D
Fig. 4b. LC-MS of cyclized octapeptin C4 analogue with Micafungin tail (R2) A.ELSD B. TIC C. UV (200 nm) D. (+)-ESI-MS
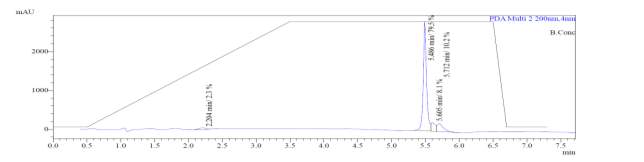
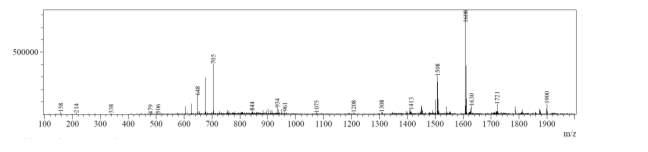
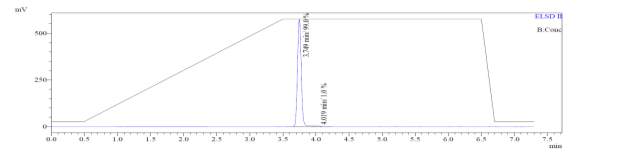
In the mass spectrum of octapeptin C4 analogue with MX-2401 tail (R3), it was evident that the most intense peak corresponded to MH+ (1562.0) and m/z 1462.0 corresponded to MH+-Boc (Fig. 4c).
A
B
C
MH+ (1562)
D
MH+-Boc (1462)
Fig. 4c. LC-MS of cyclized octapeptin C4 analogue with MX-2401 tail (R3) A.ELSD B. TIC C. UV (200 nm) D. (+)-ESI-MS
The yields of natural octapeptin C4 (R1), octapeptin C4 analogue with Micafungin tail (R2) and MX-2401 tail (R3) were 19.0% (36 mg, 100.0% purity (by UV)), 30.0% (132 mg, 80.0% purity (by UV)) and 28.0% (116 mg, 80.0% purity (by UV)) respectively. But from the LC-MS, it was evident that there were some minor impurities. Therefore, all compounds were purified again after global deprotection stage.
3.2 Characterization of compounds after global deprotection:
TFA was used to remove the Boc groups from Dab residues [33], and TIS/H2O were used to scavenge cations [34]. This was done to ensure that all the side chain protecting Boc groups are completely removed from the cyclized peptide, and global deprotection is achieved.
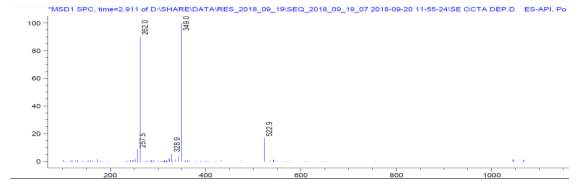
LC-MS of natural octapeptin C4 (R1) revealed that the most intense peak was MH33+ (349.0), whereas m/z 262.0 and 523.0 corresponded to MH44+ and MH22+ respectively (Fig. 5a).
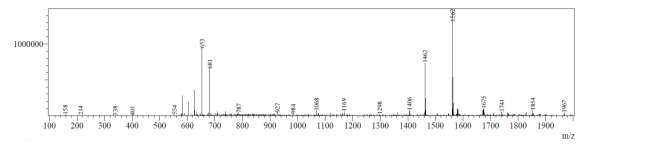
LC-MS of octapeptin C4 analogue with Micafungin tail (R2) revealed that the most intense peak was MH33+ (403.3), whereas m/z 604.0 and1208.0 corresponded to MH22+ and MH+ respectively (Fig. 5b).
LC-MS of octapeptin C4 analogue with MX-2401 tail (R3) revealed that the most intense peak was MH33+ (387.9), whereas m/z 581.0 and1162.0 corresponded to MH22+ and MH+ respectively (Fig. 5c).
MH33+ (349)
MH33+ (349.0)
MH44+ (262)
MH44+ (262)
MH22+ (523)
MH22+ (523.0)
Fig. 5a. LC-MS of fully deprotected natural octapeptin C4 (R1)
MH33+ (403)
MH22+ (604)
MH+ (1208)
Fig. 5b. LC-MS of fully deprotected octapeptin C4 analogue with Micafungin tail (R2)
MH2+2 (581)
MH3+3 (388)
MH+ (1162)
Fig. 5c. LC-MS of fully deprotected octapeptin C4 analogue with MX-2401 tail (R3)
The yields of globally deprotected natural octapeptin C4 (R1), octapeptin C4 analogue with Micafungin tail (R2) and MX-2401 tail (R3) were 95 mg, 222 mg and 193 mg respectively. However, the compounds were not free from impurities. Therefore, they were purified once again by analytical HPLC.
3.3 Characterization of compounds after HPLC:
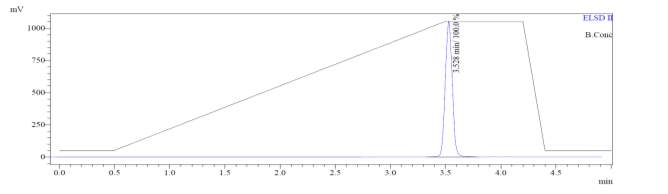
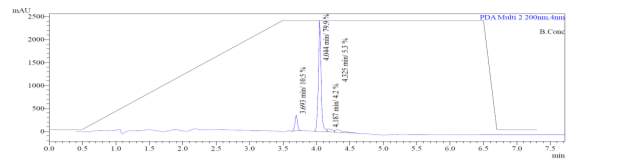
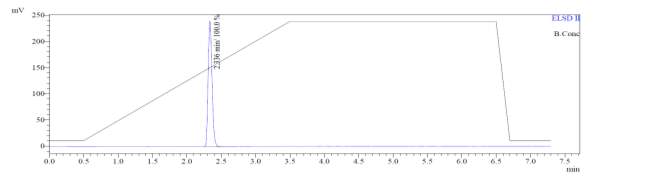
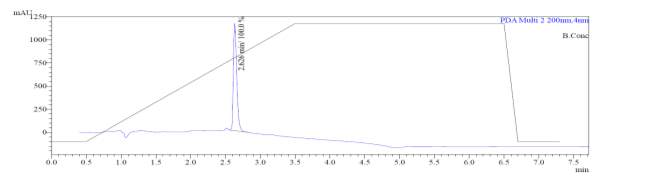
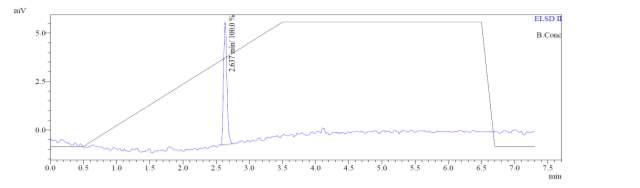
From Fig. 6a, it was evident from ELSD that there was one major, well-shaped peak at 2.336 min, with 100% purity. In TIC, a well-shaped peak was observed at 2.297 min. In case of UV, a nice peak was observed at 2.626 min, which was also 100% pure. Finally, in (+)-ESI-MS, the peak at MH33+ (348.9) corresponded to the molecular weight of the final purified compound (1044.0). The next available peaks were visible at MH22+ (523.0) and MH+ (1045.0).
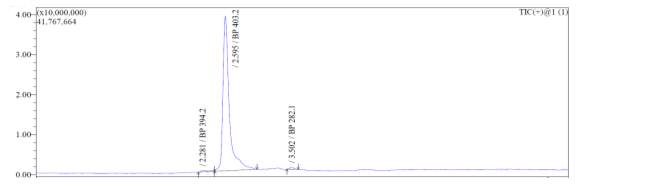
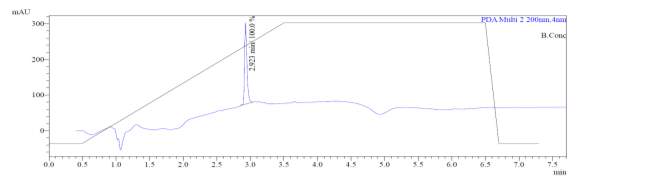
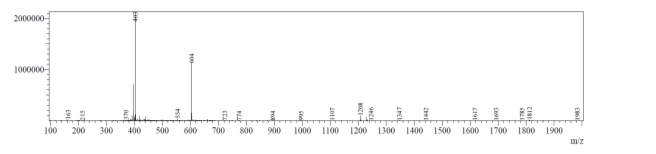
From Fig. 6b, it was evident from ELSD that there was one major, well-shaped peak at 2.637 min, with 100% purity. In TIC, a well-shaped peak was observed at 2.595 min. However, two minor peak were visible at 2.281 and 3.502 min respectively, which might have appeared due to the blank. In case of UV, a nice peak was observed at 2.923 min, which was 100% pure. Finally, in (+)-ESI-MS, the peak at MH33+ (403.2) corresponded to the molecular weight of the final purified compound (1207.0). The next available peaks were visible at MH22+ (604.0) and MH+ (1208.0).
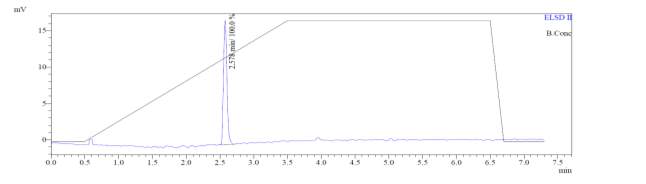
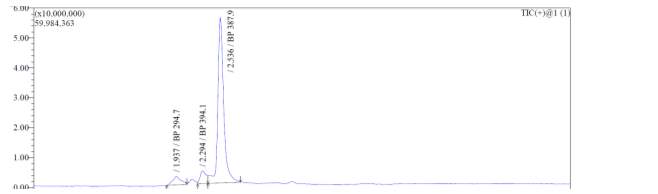
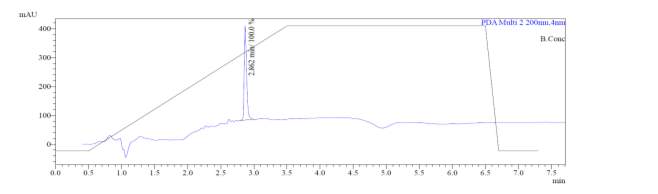
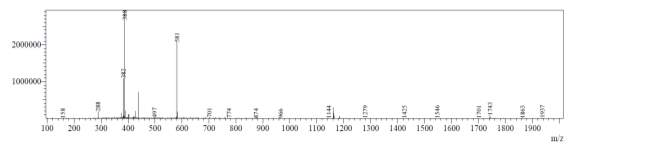
From Fig. 6c, it was evident from ELSD that there was one major, well-shaped peak at 2.578 min, with 100% purity. In TIC, a well-shaped peak was observed at 2.536 min. However, two minor peak were visible at 1.937 and 2.294 min respectively, which might have appeared due to the blank. In case of UV, a nice peak was observed at 2.862 min, which was also 100% pure. Finally, in (+)-ESI-MS, the peak at MH33+ (387.9) corresponded to the molecular weight of the final purified compound (1161.0). The next available peak was visible at MH22+ (581.0). However, the peak corresponding MH+ (1162.0) did not appear in the LC-MS, which indicate that formation of singly-charged compound for octapeptin C4 analogue with MX-2401 tail (R3) is less significant compared to double or triple charged compound.
A
B
C
MH33+ (349)
MH22+ (523)
MH+ (1045)
D
Fig. 6a. LC-MS of pure natural octapeptin C4 (R1) after HPLC A.ELSD B. TIC C. UV (200 nm) D. (+)-ESI-MS
A
B
C
MH33+ (403)
MH22+ (604)
D
MH+ (1208)
Fig. 6b. LC-MS of pure natural octapeptin C4 analogue with Micafungin tail (R2) after HPLC A.ELSD B. TIC C. UV (200 nm) D. (+)-ESI-MS
A
B
C
MH33+ (388)
MH22+ (581)
D
Fig. 6c. LC-MS of pure natural octapeptin C4 analogue with MX-2401 tail (R3) after HPLC A.ELSD B. TIC C. UV (200 nm) D. (+)-ESI-MS
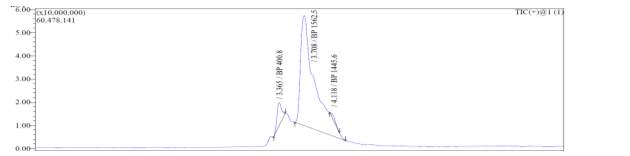
3.4 Characterization of final compounds by HRMS:
High-Resolution Mass Spectrometry (HRMS) is a useful technique to determine the exact mass of a molecule [35] and identify different compounds with identical nominal mass or elemental compositions [36]. With the introduction of time-of-flight (TOF) and other instrumental improvements, HRMS has become an authentic and reliable method for determination of exact molecular mass with highest accuracy and less chance of error [35, 36].
The HRMS exact masses (ESI microTOF-LC) for natural octapeptin C4 (R1), octapeptin C4 analogue with Micafungin tail (R2) and MX-2401 tail (R3) are presented in Table 4 as follows:
Table 4. HRMS exact mass (ESI microTOF-LC) of final compounds
| Compound with lipid group | Molecular Formula | HRMS exact mass | |
| Calculated | Observed | ||
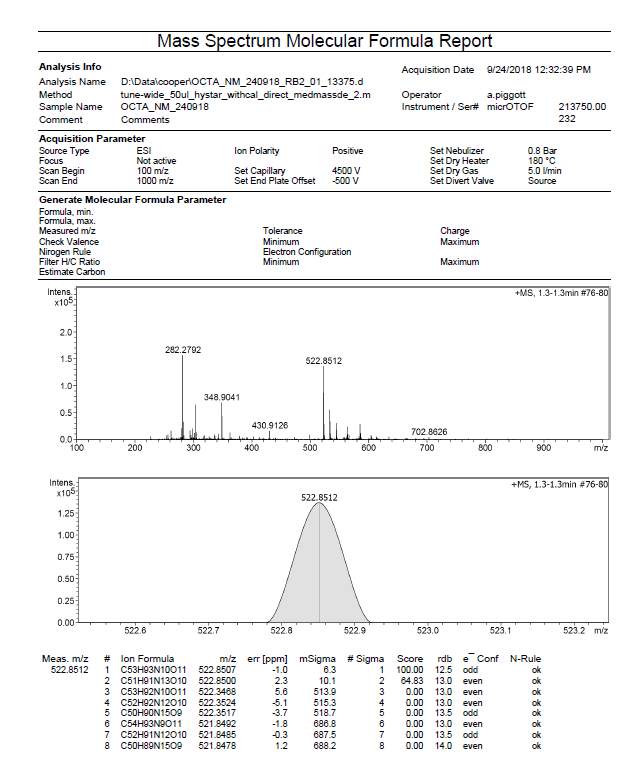
| R1 | C51H91N13O102+ | 522.8500 (MH22+) | 522.8512 |
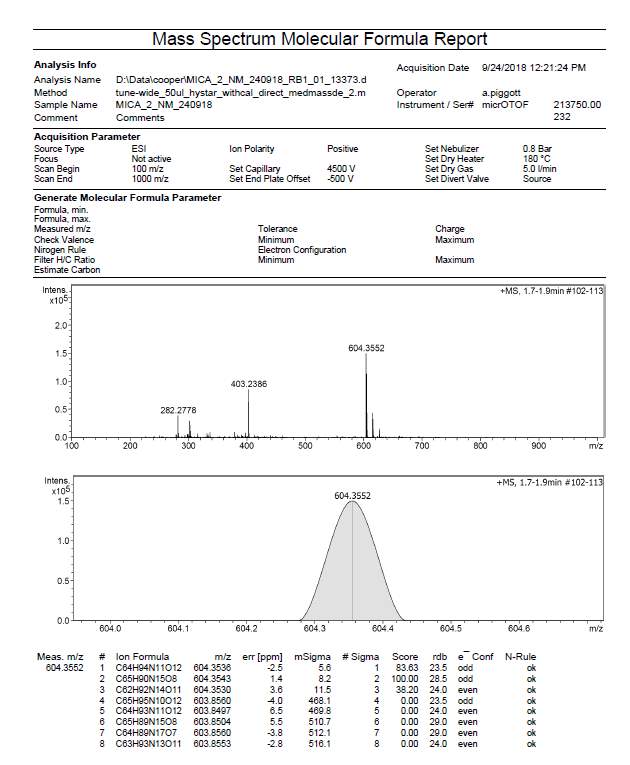
| R2 | C65H92N15O82+ | 604.3543 (MH22+) | 604.3552 |
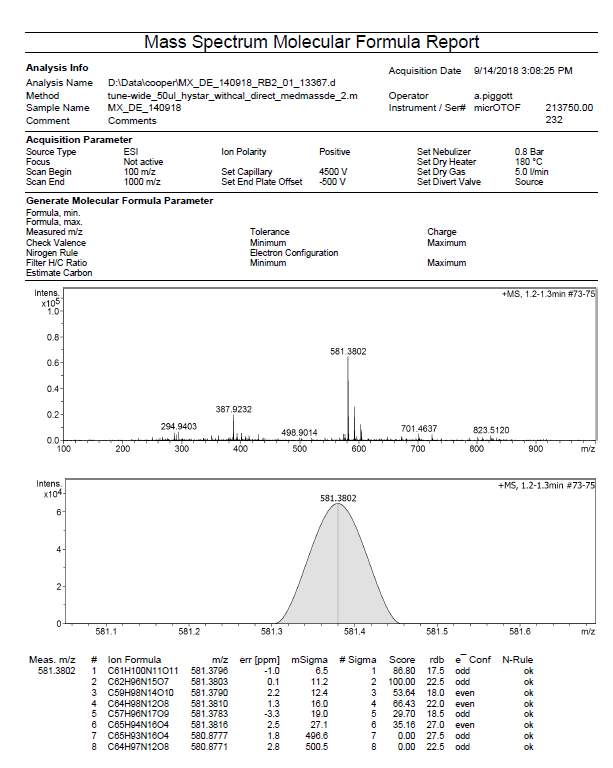
| R3 | C59H98N14O102+ | 581.3790 (MH22+) | 581.3802 |
Appendix 2. HRMS exact mass (ESI microTOF-LC) of octapeptin C4 analogue (R2)
Appendix 3. HRMS exact mass (ESI microTOF-LC) of octapeptin C4 analogue (R3)
References:
1. Vandeputte, P., Ferrari, S., Coste, A. T., Antifungal Resistance and New Strategies to Control Fungal Infections. Int J Microbiol, 2012. 3: p. 1-26.
2. Chang, Y.L., Yu, S. J., Heitman, J., Wellington, M., Chen, Y. L., New Facets of Antifungal Therapy. Virulence, 2017. 8(2): p. 222-236.
3. May, R.C., Stone, N. R. H., Wiesner, D. L., Bicanic, T., Nielsen, K., Cryptococcus: From Environmental Saprophyte to Global Pathogen. Nat Rev Microbiol, 2016. 14(2): p. 106-117.
4. Campoy, S., Adrio, J. L., Antifungals. Biochem Pharmacol, 2017. 133: p. 86-96.
5. Scorzoni, L., de Paula e Silva, A. C. A., Marcos, C. M., Assato, P. A., de Melo, W. C. M. A., de Oliviera, H. C., Costa-Orlandi, C. B., Mendes-Giannini, M. J. S., Fusco-Almeida, A. M., Antifungal Therapy: New Advances in the Understanding and Treatment of Mycosis. Front Microbiol, 2017. 8(36): p. 1-23.
6. Roemer, T., Krysan, D. J., Antifungal Drug Development: Challenges, Unmet Clinical Needs and New Approaches. Cold Spring Harb Perspect Med, 2014. 4(5): p. 1-15.
7. Perea, S., Patterson, T. F., Antifungal Resistance in Pathogenic Fungi. Clin Infect Dis, 2002. 35(9): p. 1073-1080.
8. Kathiravan, M.K., Salake, A. B., Chothe, A. S., Dudhe, P. B., Watode, R. P., Mukta, M. S., Gadhwe, S., The Biology and Chemistry of Antifungal Agents: A review. Bioorg Med Chem, 2012. 20(19): p. 5678-5698.
9. Gullo, F.P., Rossi, S. A., Sardi, J. de C. O., Teodoro, V. L. I., Mendes-Giannini, M. J. S., Fusco-Almeida, A. M., Cryptococcosis: Epidemiology, Fungal Resistance and New Alternatives for Treatment. Eur J Clin Microbiol Infect Dis, 2013. 32(11): p. 1377-1391.
10. Archibald, L.K., Tuohy, M. J., Wilson, D. A., Nwanyanwu, O., Kazembe, P. N., Tansuphasawadikul, S., Eampokalap, B., Chaovavanich, A., Reller, L. B., Jarvis, W. R., Hall, G. S., Procop, G. W., Antifungal Susceptibilities of Cryptococcus neoformans. Emerg Infect Dis, 2004. 10(1): p. 143-145.
11. Rajasingham, R., Smith, R. M., Park, B. J., Jarvis, J. N., Govender, N. P., Chiller, T. M., Denning, D. W., Loyse, A., Boulware, D. R., Global Burden of Disease of HIV-associated Cryptococcal Meningitis: An Updated Analysis. Lancet Infect Dis, 2017. 17: p. 873-881.
12. Mourad, A., Perfect, J. R., Present and Future Therapy of Cryptococcus Infections. J Fungi, 2018. 4(79): p. 1-10.
13. Chitty, J.L., Butler, M. S., Suboh, A., Edwards, D. J., Cooper, M. A., Fraser, J. A., Robertson, A. A. B., Antimicrobial Octapeptin C4 Analogues Active Against Cryptococcus Species. Antimicrob Agents Chemother, 2018. 62(2): p. 1-10.
14. Zaragoza, O., Telzak, A., Bryan, R. A., Dadachova, E., Casadevall, A., The Polysaccharide Capsule of the Pathogenic Fungus Cryptococcus neoformans Enlarges by Distal Growth and is Rearranged During Budding. Mol Microbiol, 2006. 59(1): p. 67-83.
15. Bose, I., Reese, A. J., Ory, J. J., Janbon, G., Doering, T. L., A Yeast under Cover: the Capsule of Cryptococcus neoformans. Eukaryot Cell, 2003. 2(4): p. 655-663.
16. Cherniak, R., Sundstrom, J. B., Polysaccharide Antigens of the Capsule of Cryptococcus neoformans. Infect Immun, 1994. 62(5): p. 1507-1512.
17. Davis, M.J., Eastman, A. J., Qiu, Y., Gregorka, B., Kozel, T. R., Osterholzer, J. J., Curtis, J. L., Swanson, J. A., Olszweski, M. A., Cryptococcus neoformans–Induced Macrophage Lysosome Damage Crucially Contributes to Fungal Virulence. J Immunol, 2015. 194(5): p. 2219-2231.
18. Kronstad, J., Saikia, S., Nielson, E. D., Kretschmer, M., Jung, W., Hu, G., Geddes, J. M. H., Griffiths, E. J., Choi, J., Cadiuex, B., Caza, M., Attarian, R., Adaptation of Cryptococcus neoformans to Mammalian Hosts: Integrated Regulation of Metabolism and Virulence. Eukaryot Cell, 2012. 11(2): p. 109-118.
19. Alanio, A., Vernel-Pauillac, F., Sturny-Leclere, A., Dromer, F., Cryptococcus neoformans Host Adaptation: Toward Biological Evidence of Dormancy. mBio, 2015. 6(2): p. 1-13.
20. Yang, C.L., Wang, J., Zou, L. L., Innate Immune Evasion Strategies Against Cryptococcal Meningitis Caused by Cryptococcus neoformans. Exp Ther Med, 2017. 14(6): p. 5243-5250.
21. Smith, K.D., Achan, B., Hullsiek, K. H., McDonald, T. R., Okagaki, L. H., Alhadab, A. A., Akampurira, A., Rhein, J. R., Meya, D. B., Boulware, D. R., Nielsen, K., Increased Antifungal Drug Resistance in Clinical Isolates of Cryptococcus neoformans in Uganda. Antimicrob Agents Chemother, 2015. 59: p. 7197-7204.
22. Mpoza, E., Rhein, J., Abassi, M., Emerging Fluconazole Resistance: Implications for the Management of Cryptococcal Meningitis. Med Mycol Case Rep, 2018. 19: p. 30-32.
23. Becker, B., Butler, M. S., Hansford, K. A., Gallardo-Godoy, A., Elliott, A. G., Huang, J. X., Edwards, D. J., Blaskovich, M. A. T., Cooper, M. A., Synthesis of Octapeptin C4 and Biological Profiling Aganist NDM-1 and Polymyxin Resistant Bacteria. Bioorg Med Chem Lett, 2017. 27(11): p. 2407-2409.
24. Hansen, P.R., Oddo, A., Fmoc Solid-Phase Peptide Synthesis, in Peptide Antibodies-Methods and Protocols, G. Houen, Editor. 2015, Humana Press: Statens Serum Institut, Copenhagen, Denmark. p. 33-50.
25. Merrifield, R.B., Solid Phase Peptide Synthesis, in Advances in Enzymology and Related Areas of Molecular Biology, F.F. Nord, Editor. 1969, John Wiley & Sons, Inc.: New York City. p. 221-295.
26. Fields, G.B., Noble, R. L., Solid Phase Peptide Synthesis utilizing 9-fluorenylmethoxycarbonyl Amino Acids. Int J Peptide Protein Res, 1990. 35: p. 161-214.
27. Merrifield, R.B., Solid Phase Peptide Synthesis, in The Chemistry of Polypeptides, P.G. Katsoyannis, Editor. 1973, Plenum Press: New York-London. p. 335-362.
28. Behrendt, R., White, P., Offer, J., Advances in Fmoc Solid-Phase Peptide Synthesis. J Pept Sci, 2016. 22: p. 4-27.
29. Merck. New Orthogonally Protected Lysine Derivatives. 2018 [cited 2018 16 September]; Available from: www.merckmillipore.com/novabiochem.
30. Fraser, J., MIC Laboratory Protocol. 2018, The University of Queenland.
31. ThermoFisher. Liquid Chromatography Mass Spectrometry (LC-MS) Information. 2018 [cited 2018 14 September]; Available from: https://www.thermofisher.com/au/en/home/industrial/mass-spectrometry/mass-spectrometry-learning-center/liquid-chromatography-mass-spectrometry-lc-ms-information.html.
32. Chemyx. An overview of HPLC, MS, and LC-MS. 2018 [cited 2018 14 September]; Available from: https://www.chemyx.com/support/knowledge-base/applications/basic-principles-hplc-ms-lc-ms/.
33. Guy, C.A., Fields, G. B., Trifluoroacetic Acid Cleavage and Deprotection of Resin-bound Peptides Following Synthesis by Fmoc Chemistry. Methods Enzymol, 1997. 289: p. 67-83.
34. Pearson, D.A., Blanchette, M., Baker, M. L., Guindon, C. A., Trialkylsilanes as Scavengers For The Trifluoroacetic acid Deblocking of Protecting Groups in Peptide Synthesis. Tetrahedron Lett, 1989. 30(21): p. 2739-2742.
35. Pleil, J.D., Isaacs, K. K., High Resolution Mass Spectrometry: Basic Principles for Using Exact Mass and Mass Defect for Discovery Analysis of Organic Molecules in Blood, Breath, Urine and Environmental Media. J Breath Res, 2016. 10: p. 1-10.
36. Stock, N.L., Introducing Graduate Students to High-Resolution Mass Spectrometry (HRMS) Using a Hands-On Approach. J Chem Educ, 2017. 94: p. 1978-1982.
Cite This Work
To export a reference to this article please select a referencing stye below:
Reference Copied to Clipboard.
Reference Copied to Clipboard.
Reference Copied to Clipboard.
Reference Copied to Clipboard.
Reference Copied to Clipboard.
Reference Copied to Clipboard.
Reference Copied to Clipboard.
Related Services
View all


Related Content
All TagsContent relating to: "Pharmacology"
Pharmacology involves the study of drugs and how they affect the body. A pharmacologist contributes to drug development by researching and testing how the body reacts to medication, and whether the medication can have a positive impact on the body in terms of fighting illness and disease.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: