The Mode of Action of Artemisinin and its Cellular and Molecular Bases of Resistance
Info: 4914 words (20 pages) Dissertation
Published: 10th Dec 2019
Tagged: PharmacologyBiomedical Science
Introduction to Special Study Project
Malaria is a debilitating and life-threatening disease that affects some 91 countries worldwide. This protozoan infection caused 429,000 deaths in 2015 alone, of which 303,000 were children leading to an estimate that this disease kills a child every 2 minutes. (WHO, 2016)
 World Malaria Report 2016 reveals that the current global management of this disease is successful, as suggested by declining malarial prevalence and mortality rates since 2000. This is indicated by tables 1 and 2 below (WHO, 2016). Furthermore, this report attributes the relative success of malaria management to factors such as: boosted research funding, greater access to malarial therapies and a significant rise in the utility of insecticide-treated mosquito nets (ITN) and indoor residual sprays (IRS). As a result, ambitious initiatives were launched that aim to eliminate malaria, such as the Global Technical Strategy for Malaria 2016-2030 (WHO, 2015) which targets a 90% reduction in prevalence and mortality rates of malaria by 2030, and the Sustainable Development Goals (Nino, 2015) that aim to completely eradicate this disease by 2030.
World Malaria Report 2016 reveals that the current global management of this disease is successful, as suggested by declining malarial prevalence and mortality rates since 2000. This is indicated by tables 1 and 2 below (WHO, 2016). Furthermore, this report attributes the relative success of malaria management to factors such as: boosted research funding, greater access to malarial therapies and a significant rise in the utility of insecticide-treated mosquito nets (ITN) and indoor residual sprays (IRS). As a result, ambitious initiatives were launched that aim to eliminate malaria, such as the Global Technical Strategy for Malaria 2016-2030 (WHO, 2015) which targets a 90% reduction in prevalence and mortality rates of malaria by 2030, and the Sustainable Development Goals (Nino, 2015) that aim to completely eradicate this disease by 2030.
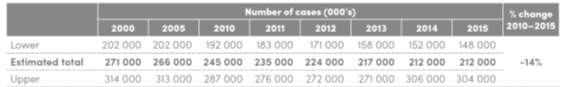
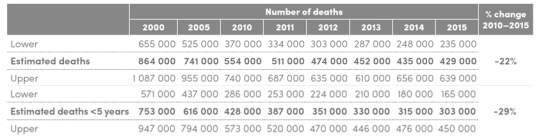 Table 1 indicates estimations of malaria cases between 2000 to 2015. This suggests that the global malarial prevalence is decreasing, as illustrated by the fact that the estimated total number of cases of malaria in 2000 was 271,000,000, which declined by 22% to 212,000,000 cases in 2015.
Table 1 indicates estimations of malaria cases between 2000 to 2015. This suggests that the global malarial prevalence is decreasing, as illustrated by the fact that the estimated total number of cases of malaria in 2000 was 271,000,000, which declined by 22% to 212,000,000 cases in 2015.
Table 2 indicates number of deaths caused by malaria between 2000 to 2015. This suggests that the global malarial mortality is decreasing, as shown by the comparison where the number of deaths in 2000 was 864,000 which declined by 50% to 429,000 in 2015. Furthermore, the infantile mortality in infants under 5 years of age is also decreasing, as there were 753,000 infant deaths in 2000 which declined by 60% to 303,000 in 2015.
However, while malaria may be declining in-terms of prevalence and mortality, it is growing as a threat to direct global health. This is because of experimental evidence that suggest parasitic resistance to the main anti-malarial therapies, such as to those that contain artemisinin, sulphadoxine-pyrimethamine and chloroquine. Furthermore, studies have also detected anopheline resistance to pyrethroids (main insecticide utilised in ITN). As a result, the relative success and effectiveness of current treatments are decreasing with time, whereby the aims of GTS and SDG to eliminate malaria are likely to be compromised (Walker et al., 2014).
In this special study project, I will begin by briefly outlining the biological basis of malaria, which can aid in understanding how many of the anti-malarial agents work. Following this, I will focus in-depth on artemisinin and its derivatives, as they form the primary components of most frontline anti-malarial therapies utilised in the current day. This involves discussing their mode of action and highlighting the cellular and molecular bases of parasitic resistance to this drug.
The Biological Basis of Malaria
Malaria arises following a protozoan infection by sporozoan species of the plasmodium genus, which include P. falciparum (most virulent), P. vivax, P. ovale, P. malariae and P. knowlesi. These parasites are most commonly transmitted via the bite of an infected female Anopheles mosquito, as this permits direct transfer of sporozoites into the human host. Other common modes of transmission include infected blood transfusion, infected needle use and congenital transmission.
Following invasion, the injected sporozoites enter the hypnozoite stage where they invade hepatocytes to undergo exoerythrocytic schizogony (asexual reproduction outside erythrocytic cells). However, some P. vivax and P. ovale hypnozoites become latent instead, whereby they do not reproduce for a period of time until after they are reactivated. Eventually, these cells rupture to release daughter parasites (merozoites) into blood, where the erythrocytic stage occurs as merozoites invade erythrocytes to mediate the ring stage. This leads onto the trophozoite stage and later the schizont stage, where the merozoites pierce the erythrocyte plasma membrane to obtain entry into the cell cytosol. The parasites now exist in parasitophorous vacuoles where they grow and proliferate. (Schäfer, 2014a)
During this period, parasites form food vacuoles where haemoglobin is taken-up and catalysed via protease enzymes into amino acids and toxic ferriprotoporphyrin (FPIX). While the amino acids are catabolised into proteins for either merozoite synthesis, cytoadherence or immune system evasion, the FPIX is non-enzymatically detoxified into non-toxic hemozoin via biocrystallisation to prevent the generation of reaction oxygen species (Schäfer, 2014a).
Eventually, the parasite will have replicated as maximally as the erythrocyte will accommodate, after which the cell ruptures to release merozoites and other cell contents into blood. The merozoites infect other erythrocytes to repeat the cycle, while the lysed erythrocyte debris coordinates the cytokine-driven immune responses that mediate most malarial symptoms (White et al., 2014).
Artemisinin as an Anti-Malarial Agent
The dried herbs of a year-old Artemisia annua (sweet wormwood or annual mugwort) has been a traditional Chinese remedy for febrile conditions for over 1600 years. Its active compound is artemisinin, an insoluble sesquiterpene lactone that contains the endoperoxide bridge (1,2,4-trioxane). Artemisinin structure is shown in the figure below (O’Neill et al., 2010). This was first isolated in 1972 by Nobel Laureate: Youyou Tu while she was head of Project 523, via the steps of degradation and X-ray crystallography. (Miller and Su, 2011)
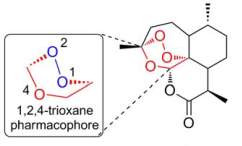
Structural representation of artemisinin, which is a sesquiterpene lactone and so consists of three isoprene units and a lactone ring. Particular focus is placed on the endoperoxide bridge, which is the 1,2,4-trioxane structure (O’Neill et al., 2010).
However, the direct use of sweet wormwood as an anti-malarial therapy is widely prohibited. This is because these herbs possess a low artemisinin content, and so their use would generate insufficient therapeutic effects and encourage parasitic resistance by natural selection. Therefore, artemisinin is instead obtained in sufficient concentrations by either: chemical synthesis via complex pathways involving extraction from sweet wormwood, or fermentative production of artemisinic acid followed by reduction and esterification (Schäfer, 2014b).
Artemisinin is a potent anti-malarial agent that is limited by low bioavailability and a short half-life. Therefore, synthetic derivatives that overcome these pharmacokinetic limitations are used instead, so much so that they are the frontline treatment for malaria. They are usually offered alongside other anti-malarial agents in artemisinin-based combination therapy (ACT), which increases therapeutic effect and reduces risk of developing artemisinin-resistance.
Common artemisinin derivatives include artesunate, artemether, arteether, dihydroartemisinin (active biological form of artemisinin) and artemotil. These derivatives possess chemical modifications that increase water and lipid solubility, which in-turn improves pharmacokinetic properties to increase bioavailability and half-life (Schäfer, 2014b).
Furthermore, the 7 chiral centres of artemisinin result in 128 different optical isomers, whereby certain isomers generate the desired biological effect while others do not. Therefore, these steric properties suggest that artemisinin’s mode of action comprises of complementary binding onto a molecular target, such as protein.
Mode of Artemisinin Action: Artemisinin Bioactivation
The precise mode of artemisinin action is unclear. However, Meshnik et al. in 1991 first described the widely accepted idea that 1,2,4-trioxane (endoperoxide bridge) of artemisinin acts as the pharmacophore when bioactivated by parasite-derived haem iron (Meshnick et al., 1991).
Therefore, artemisinin is converted into dihydroartemisinin when the Fe2+ released from haemoglobin degradation interacts with 1,2,4-trioxane. This is believed to occur via either: the reductive scission model where Fe2+ binds to 1,2,4-trioxane to allow reductive cleavage, or the open peroxide model where Fe2+ protonates 1,2,4-trioxane to allow heterolytic cleavage instead. However, both models give rise to bio-radicals that induce parasitic damage, which explains the therapeutic benefits of artemisinin (O’Neill et al., 2010). The detailed mechanism for each model is illustrated in the figure below (O’Neill et al., 2010).
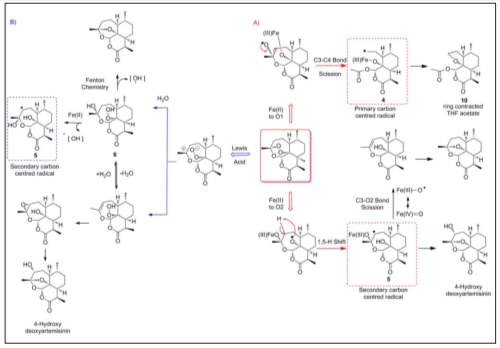
Mechanisms of the reductive scission and open peroxide models (O’Neill et al., 2010), whereby:
A) Mechanism of reductive scission where Fe2+ reduces the artemisinin pharmacophore to allow reductive cleavage that forms oxygen radicals that rearrange into carbon radicals.
B) Mechanism of open peroxide where Fe2+ protonates the artemisinin pharmacophore, as it acts as a Lewis acid, to allow heterolytic cleavage that forms an unsaturated hydroperoxide that then undergoes the Fenton reaction to form hydroxyl radicals.
Mode of Artemisinin Action: Mechanism of Parasite Destruction
While bio-radicals are understood to destroy malarial parasites, the mechanism of this destruction is unclear. However, there are numerous propositions that attempt to explain this phenomenon.
Recent evidence indicates that bio-radicals inhibit P. falciparum phosphatidylinositol-3-kinase (pfPI3K) to indirectly reduce phosphatidylinositol-3-phosphate (PI3P) synthesis. As a result, PI3P-dependent signalling is reduced, which mediates parasitic dysfunction by incorrectly directing proteins destined for host erythrocyte away from the endoplasmic reticulum (ER) and into the parasitophorous vacuole (default secretory pathway) (Mbengue et al., 2015).
Evidence for this includes biochemical analyses that reveal PI3P production is quickly blocked following dihydroartemisinin administration, but is resumed after drug wash-out. Also, this could not be replicated in deoxyartemisinin, which is an artemisinin-derivative that does not possess 1,2,4-trioxane. Therefore, this indicates the importance of the endoperoxide bridge in this interaction. Furthermore, structural analysis show that dihydroartemisinin displays excellent complementarity with hydrophobic regions of pfPI3K, which explains why nanomolar concentrations are capable of inhibiting PI3P synthesis so well.
The precise role of PI3P is largely unknown, however, it is understood that within parasitic ER the PI3P binds with high affinity onto host-targeting (HT) signals on proteins destined for the erythrocyte. Therefore, it is presumed that this lipid plays a critical role in exporting proteins (Bhattacharjee et al., 2012).
One such suggestion is the radicals alkylate FPIX to reduce detoxification efficiency. As a result, toxic FPIX levels rise to form oxygen radicals that damage parasites by inducing oxidative stress (Robert et al., 2006). This is supported by the formation of covalent FPIX-artemisinin adducts when FPIX is incubated with artemisinin, as this can only occur following FPIX alkylation (Robert et al., 2006). Furthermore, radiolabelled artemisinin was found to readily alkylate in-vitro haemoproteins such as catalase, cytochrome C and haemoglobin (Ying-Zi et al., 1994).
Alternatively, they may alkylate parasitic proteins to reduce parasite functionality. This explains artemisinin’s steric properties as only certain isomers can bind complementarily to protein targets to create an effect while the others cannot (O’Neill et al., 2010). Evidence for this is provided by radiographs that show radiolabelled arteether, dihydroartemisinin and arteflene bound to proteins in infected erythrocytes but not healthy erythrocytes. Therefore, this suggests that artemisinin binds to parasitic proteins to mediate an effect rather than erythrocytic proteins (Asawamahasakda et al., 1994).
Other viable proposals include: oxidation of food vacuole membranes (Hartwig et al., 2009), inhibition of parasitic SERCA (Pulcini et al., 2013), and disruption of disulphide reductase (Haynes et al., 2011).
Artemisinin Resistance
For the most part, artemisinin and its derivatives are extremely effective against malaria, particularly that induced by P. falciparum. However, studies from as far back as 2006 have indicated artemisinin resistance in the Greater Mekong Sub-Region of Southeast Asia, which was first detected along the Thai-Cambodian border but has since spread to other areas in this sub-region too (CDC, 2017).
An open-label study coordinated by Dondorp et al. in 2009 indicated P. falciparum clearance following artesunate monotherapy or artesunate-mefloquine therapy (ACT) was significantly slower in Pailin (western Cambodia) than Wang Pha (north-western Thailand). This supports the idea of parasitic resistance against artemisinin, particularly as this drug has been the frontline anti-malarial agent in Pailin for over 35 years but was only introduced in Wang Pha in 1994, which implies that there ought to be a greater prominence of resistance in Pailin than in Wang Pha.
The sample comprised 40 falciparum malaria patients in each location that were assigned to either: artesunate monotherapy or artesunate-mefloquine therapy. Both these groups were hospitalised while they received designated therapy for a duration of 7 days. Meanwhile, P. falciparum susceptibility and resistance markers were analysed to monitor effective of treatments provided.
The results found that the median parasite clearance time across both groups was 84 hours in Pailin and 48 hours in Wang Pha. Therefore, the overall effectiveness of artemisinin-based therapy is greater in Wang Pha than Pailin. The experimental data is shown in the graph below (Dondorp et al., 2009).
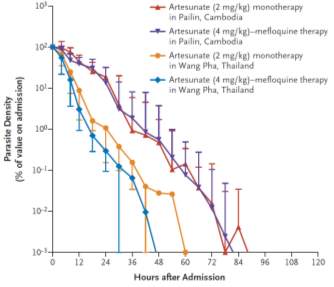
Graph derived from the open-label study coordinated by Dondorp et al., which maps Parasite Density (% of value on admission) against hours after admission. This indicates that artesunate monotherapy and artesunate-mefloquine therapy (ACT) are effective cures for malaria in both Pailin and Wang Pha (shown by a decline in parasite density across all groups to 10−3%), however, artesunate-mefloquine therapy appears to have a greater therapeutic effect in both locations as this reaches 10−3% parasite density quickest. Furthermore, the time taken to reach 10−3% varies between the two locations, whereby the average time taken in Wang Pha is 54 hours while in Pailin is 84 hours. Therefore, this suggests there is a greater prominence of parasitic resistance in Pailin where artemisinin has been used routinely for over 35 years, than Wang Pha where this was introduced in just 1994 (Dondorp et al., 2009).
Cellular and Molecular Basis of Artemisinin Resistance
Artemisinin resistance is attributed to alterations in the parasitic genome via natural selection, whereby the selected gene encode products that counteract drug efficacy. The precise gene loci responsible for resistant P. falciparum is unknown, but numerous genome-wide association studies (GWAS) have outlined potential candidates such as: chromosome 13 SNPs – MAL13-1718319 and MAL13-1719976 (Takala-Harrison et al., 2013) or mutations in genes encoding lipoate synthase, aminomethyltransferase and hsp70 (Cheeseman et al., 2012).
EARLY RING STAGE
However, P. falciparum Kelch13 (pfK13) gene mutations are widely considered as the likely causal factor for artemisinin resistance. These exist as 21 possible variations, which frequently manifest as C580Y (most common), Y493H, R539T or I543T, (Singh et al., 2016). Evidence supporting this is provided by a study coordinated by Ghorbal et al. in 2014, where artemisinin-sensitive parasites were transfected in-vitro with mutated pfK13 genes via the CRISPR-Cas9 system. Within three weeks of transfection, these parasites developed artemisinin-resistance, whereby the RSA0–3 h survival assay revealed that ring-stage parasite survival had increased by 13.5% (Ghorbal et al., 2014).
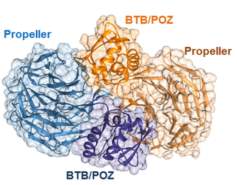 The pfK13 protein comprises of 726 amino acid residues containing two domains: Kelch propeller which allows substrate-binding, and BTB/POZ which promotes dimerisation. The physiological function of pfK13 is poorly understood, however, it is accepted that it is a homomeric structure that ubiquitylates substrate prior to degradation by non-specific proteolysis. Importantly, this protein degrades P. falciparum phosphatidylinositol-3-kinase (pfPI3K), which is an enzyme that catalyses the formation of phosphatidylinositol-3-phosphate (PI3P) (Singh et al., 2016). The structure of the pfK13 homodimer is illustrated below (Singh et al., 2016).
The pfK13 protein comprises of 726 amino acid residues containing two domains: Kelch propeller which allows substrate-binding, and BTB/POZ which promotes dimerisation. The physiological function of pfK13 is poorly understood, however, it is accepted that it is a homomeric structure that ubiquitylates substrate prior to degradation by non-specific proteolysis. Importantly, this protein degrades P. falciparum phosphatidylinositol-3-kinase (pfPI3K), which is an enzyme that catalyses the formation of phosphatidylinositol-3-phosphate (PI3P) (Singh et al., 2016). The structure of the pfK13 homodimer is illustrated below (Singh et al., 2016).
3D crystal structure of the pfK13 homodimer as determined by the Structural Genomics Consortium (Singh et al., 2016). This homomeric proteins comprises of two pfK13 proteins (one is blue, other is orange) that dimerise via the BTB/POZ domains. Meanwhile, the Kelch propeller domains form the substrate binding site.
Most resistance mutations of pfK13 (including C580Y) induce resistance by altering the structure of the Kelch propeller, leading to a reduced ability to bind substrate. As a result, pfPI3K is inadequately degraded due to insufficient ubiquitylation to allow PI3P levels to rise. This mediates artemisinin resistance by overcoming artemisinin-induced inhibition of PI3P-dependent signalling, and so corrects the directing of protein export from the ER, past the parasitophorous vacuole and into the cytosol and plasma membrane of the erythrocyte to coordinate the appropriate erythrocytic changes that aid in parasitic survival.
However, immense ambiguity over artemisinin’s mode of action means there is currently no definite theory for artemisinin resistance.
Outlook for Artemisinin Resistance
Parasitic resistance to other anti-malarial agents similarly originated along the Thai-Cambodian border before spreading into Asia and Africa (Walker et al., 2014). This includes P. falciparum resistance to: chloroquine due to pfCRT gene mutations (Ecker et al., 2012) and sulphadoxine-pyrimethamine due to pfDHPS and pfDHRF gene mutations (Okell et al., 2017). Therefore, this proposes that artemisinin may display a similar trend in the future, whereby it may become ineffective with time due to an increased spread of resistance. This has already been indicated in numerous research findings, such as that in the graph below (Dondorp et al., 2011).
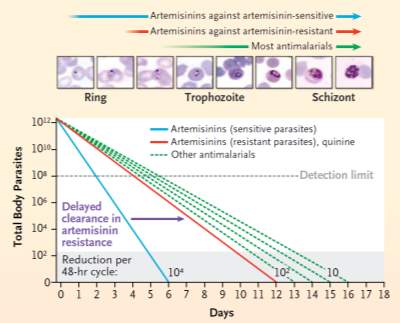
Graph mapping the total body malarial parasites against the number of days. This indicates that while all anti-malarial agents have potential to cure malaria, the rates at which they achieve this is very different. For example, most anti-malarial agents cure malaria between 13-16 days after admission, while artemisinin cures malaria in 6 days if the parasite is artemisinin-sensitive or 12 days if the parasite is artemis2inin-resistant. As a result, this illustrates that the emergence of artemisinin resistant parasites may render artemisinin as ineffective as the other anti-malarial agents, which in practice would be disastrous in malaria management (Dondorp et al., 2011).
However, widespread artemisinin resistance would have catastrophic effects on global malarial management, leading to a significant rise mortality rates. As a result, globally-derived initiatives for artemisinin-resistance elimination in the Greater Mekong sub-region were launched (Canavati et al., 2016). These programs target artemisinin-resistant parasites in numerous ways, such as by encouraging: introduction of artesunate-pyronaridine ACT, direct observed therapy (DOT), malaria education and removal of artemisinin monotherapies and counterfeit anti-malarial agents (Dondorp et al., 2011).
Reference List
Asawamahasakda, W., Ittarat, I., Pu, Y.M., Ziffer, H., Meshnick, S.R., 1994. Reaction of antimalarial endoperoxides with specific parasite proteins. Antimicrob. Agents Chemother. 38, 1854–1858.
Bhattacharjee, S., Stahelin, R.V., Speicher, K.D., Speicher, D.W., Haldar, K., 2012. Endoplasmic Reticulum PI(3)P lipid binding targets malaria proteins to the host cell. Cell 148, 201–212. https://doi.org/10.1016/j.cell.2011.10.051
Canavati, S.E., Lawford, H.L.S., Fatunmbi, B.S., Lek, D., Top-Samphor, N., Leang, R., Dondorp, A.M., Huy, R., Kazadi, W.M., 2016. Establishing research priorities for malaria elimination in the context of the emergency response to artemisinin resistance framework-the Cambodian approach. Malar. J. 15, 120. https://doi.org/10.1186/s12936-016-1117-9
CDC, 2017. CDC – Malaria [WWW Document]. URL https://www.cdc.gov/malaria/ (accessed 11.1.17).
Cheeseman, I.H., Miller, B.A., Nair, S., Nkhoma, S., Tan, A., Tan, J.C., Saai, S.A., Phyo, A.P., Moo, C.L., Lwin, K.M., McGready, R., Ashley, E., Imwong, M., Stepniewska, K., Yi, P., Dondorp, A.M., Mayxay, M., Newton, P.N., White, N.J., Nosten, F., Ferdig, M.T., Anderson, T.J.C., 2012. A major genome region underlying artemisinin resistance in malaria. Science 336, 79–82. https://doi.org/10.1126/science.1215966
Dondorp, A.M., Fairhurst, R.M., Slutsker, L., MacArthur, J.R., M.D., J.G.B., Guerin, P.J., Wellems, T.E., Ringwald, P., Newman, R.D., Plowe, C.V., 2011. The Threat of Artemisinin-Resistant Malaria. N. Engl. J. Med. 365, 1073–1075. https://doi.org/10.1056/NEJMp1108322
Dondorp, A.M., Nosten, F., Yi, P., Das, D., Phyo, A.P., Tarning, J., Lwin, K.M., Ariey, F., Hanpithakpong, W., Lee, S.J., Ringwald, P., Silamut, K., Imwong, M., Chotivanich, K., Lim, P., Herdman, T., An, S.S., Yeung, S., Singhasivanon, P., Day, N.P.J., Lindegardh, N., Socheat, D., White, N.J., 2009. Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 361, 455–467. https://doi.org/10.1056/NEJMoa0808859
Ecker, A., Lehane, A.M., Clain, J., Fidock, D.A., 2012. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 28, 504–514. https://doi.org/10.1016/j.pt.2012.08.002
Ghorbal, M., Gorman, M., Macpherson, C.R., Martins, R.M., Scherf, A., Lopez-Rubio, J.-J., 2014. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat. Biotechnol. 32, nbt.2925. https://doi.org/10.1038/nbt.2925
Hartwig, C.L., Rosenthal, A.S., D’Angelo, J., Griffin, C.E., Posner, G.H., Cooper, R.A., 2009. Accumulation of artemisinin trioxane derivatives within neutral lipids of Plasmodium falciparum malaria parasites is endoperoxide-dependent. Biochem. Pharmacol. 77, 322–336. https://doi.org/10.1016/j.bcp.2008.10.015
Haynes, R.K., Cheu, K.-W., Li, K.-Y., Tang, M.M.-K., Wong, H.-N., Chen, M.-J., Guo, Z.-F., Guo, Z.-H., Coghi, P., Monti, D., 2011. A Partial Convergence in Action of Methylene Blue and Artemisinins: Antagonism with Chloroquine, a Reversal with Verapamil, and an Insight into the Antimalarial Activity of Chloroquine. ChemMedChem 6, 1603–1615. https://doi.org/10.1002/cmdc.201100184
Mbengue, A., Bhattacharjee, S., Pandharkar, T., Liu, H., Estiu, G., Stahelin, R.V., Rizk, S.S., Njimoh, D.L., Ryan, Y., Chotivanich, K., Nguon, C., Ghorbal, M., Lopez-Rubio, J.-J., Pfrender, M., Emrich, S., Mohandas, N., Dondorp, A.M., Wiest, O., Haldar, K., 2015. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520, nature14412. https://doi.org/10.1038/nature14412
Meshnick, S.R., Thomas, A., Ranz, A., Xu, C.M., Pan, H.Z., 1991. Artemisinin (qinghaosu): the role of intracellular hemin in its mechanism of antimalarial action. Mol. Biochem. Parasitol. 49, 181–189.
Miller, L.H., Su, X., 2011. Artemisinin: Discovery from the Chinese Herbal Garden. Cell 146, 855–858. https://doi.org/10.1016/j.cell.2011.08.024
Nino, F.S., 2015. Sustainable development goals – United Nations. U. N. Sustain. Dev.
Okell, L.C., Griffin, J.T., Roper, C., 2017. Mapping sulphadoxine-pyrimethamine-resistant Plasmodium falciparum malaria in infected humans and in parasite populations in Africa. Sci. Rep. 7, 7389. https://doi.org/10.1038/s41598-017-06708-9
O’Neill, P.M., Barton, V.E., Ward, S.A., 2010. The Molecular Mechanism of Action of Artemisinin—The Debate Continues. Molecules 15, 1705–1721. https://doi.org/10.3390/molecules15031705
Pulcini, S., Staines, H.M., Pittman, J.K., Slavic, K., Doerig, C., Halbert, J., Tewari, R., Shah, F., Avery, M.A., Haynes, R.K., Krishna, S., 2013. Expression in Yeast Links Field Polymorphisms in PfATP6 to in Vitro Artemisinin Resistance and Identifies New Inhibitor Classes. J. Infect. Dis. 208, 468–478. https://doi.org/10.1093/infdis/jit171
Robert, A., Bonduelle, C., Laurent, S.A.-L., Meunier, B., 2006. Heme alkylation by artemisinin and trioxaquines. J. Phys. Org. Chem. 19, 562–569. https://doi.org/10.1002/poc.1059
Schäfer, B., 2014a. Artemisinin. Chem. Unserer Zeit 48, 134–145. https://doi.org/10.1002/ciuz.201400645
Schäfer, B., 2014b. Artemisinin. Chem. Unserer Zeit 48, 216–225. https://doi.org/10.1002/ciuz.201400652
Singh, G.P., Goel, P., Sharma, A., 2016. Structural mapping of Kelch13 mutations associated with artemisinin resistance in malaria. J. Struct. Funct. Genomics 17, 51–56. https://doi.org/10.1007/s10969-016-9205-1
Takala-Harrison, S., Clark, T.G., Jacob, C.G., Cummings, M.P., Miotto, O., Dondorp, A.M., Fukuda, M.M., Nosten, F., Noedl, H., Imwong, M., Bethell, D., Se, Y., Lon, C., Tyner, S.D., Saunders, D.L., Socheat, D., Ariey, F., Phyo, A.P., Starzengruber, P., Fuehrer, H.-P., Swoboda, P., Stepniewska, K., Flegg, J., Arze, C., Cerqueira, G.C., Silva, J.C., Ricklefs, S.M., Porcella, S.F., Stephens, R.M., Adams, M., Kenefic, L.J., Campino, S., Auburn, S., MacInnis, B., Kwiatkowski, D.P., Su, X., White, N.J., Ringwald, P., Plowe, C.V., 2013. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc. Natl. Acad. Sci. U. S. A. 110, 240–245. https://doi.org/10.1073/pnas.1211205110
Walker, N.F., Nadjm, B., Whitty, C.J.M., 2014. Malaria. Medicine (Baltimore), Tropical Infections 42, 100–106. https://doi.org/10.1016/j.mpmed.2013.11.011
White, N.J., Pukrittayakamee, S., Hien, T.T., Faiz, M.A., Mokuolu, O.A., Dondorp, A.M., 2014. Malaria. The Lancet 383, 723–735. https://doi.org/10.1016/S0140-6736(13)60024-0
WHO, 2016. WHO | World Malaria Report 2016 [WWW Document]. WHO. URL http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/ (accessed 11.1.17).
WHO, 2015. WHO | Global Technical Strategy for Malaria 2016–2030 [WWW Document]. WHO. URL http://www.who.int/malaria/areas/global_technical_strategy/en/ (accessed 11.1.17).
WHO, n.d. WHO | World Malaria Report 2016 [WWW Document]. WHO. URL http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/ (accessed 10.31.17).
Ying-Zi, Y., Little, B., Meshnick, S.R., 1994. Alkylation of proteins by artemisinin. Biochem. Pharmacol. 48, 569–573. https://doi.org/10.1016/0006-2952(94)90287-9
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biomedical Science"
Biomedical Science focuses on how cells, organs and systems function in the human body and underpins much of modern medicine. Biomedical Science applies parts of natural and/or formal sciences to help develop advances in healthcare.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: