Nanoparticle Formulations for Sustained Delivery and Brain Targeting of Neuropeptide Y
Info: 9324 words (37 pages) Dissertation
Published: 14th Feb 2022
Abstract
Neuropeptide Y (NPY) is a potential candidate for the treatment of epilepsy and other neurodegenerative diseases as previous studies have documented that the brain NPY system has anti-seizure activity. With all these advantages of NPY, it cannot readily pass the blood-brain barrier as only very small lipophilic molecules can cross the blood-brain barrier independently. To generate a brain penetrant and sustained release formulation of NPY, in this study, we fabricated polymeric nanoparticle formulations using poly(lactic-co-glycolic acid) (PLGA) or bovine serum albumin (BSA) as the base material and conjugating each material to either transferrin or rabies virus glycoprotein (RVG) as brain targeting ligands. All four formulations were characterized in vitro for size, zeta potential, encapsulation efficiency, and release profiles. All formulations showed slightly negative charges and sizes ranging from 137 to 267.4 nm in diameter, with RVG-conjugated BSA nanoparticles exhibiting the smallest sizes. No formulation was found to be immunogenic or cytotoxic. The encapsulation efficiency was ≥ 75% for all nanoparticle formulations and release studies demonstrated that all formulations have a sustained release profile. All BSA formulations exhibited a faster initial burst of release compared to PLGA particles, in addition to later sustained release. This initial burse release would be helpful for clinical dosing as therapeutic effects could be quickly established. Additional sustained release can be helpful to maintain the therapeutic effects. In vitro data including size and release profile indicate that RVG-conjugated BSA nanoparticles are the most favorable formulation for brain delivery of Neuropeptide Y.
Keywords: Autism, Blood-brain barrier (BBB), Brain delivery, Neuropeptide Y, Nanoparticle
Introduction
There is an urgent need to development of new treatments for diseases of the central nervous system (CNS) as it is a pressing public health concern (Broderick, Schweitzer, & Wszolek, 2009; Herrmann, Chau, Kircanski, & Lanctot, 2011; Misra, Ganesh, Shahiwala, & Shah, 2003; Strecker & Schwarz, 2008). Delivering beneficial therapies, such as neuropeptides across the blood-brain barrier (BBB) can be a potential way to treat neurological diseases. Neuropeptides are commonly co-released with neurotransmitters to modulate neurotransmission and are small protein-like molecules. Previous studies have shown that neuropeptides can be potential therapeutics for CNS disorders as several animal studies demonstrated profound efficacy in treating a number of disease states (Duarte-Neves, Pereira de Almeida, & Cavadas, 2016). Although there are many neuropeptides with potential therapeutic benefits, neuropeptide Y (NPY) is of particular interest. NPY is a 36 amino-acid neuropeptide. NPY is found to be involved in various physiological and homeostatic processes in both the central and peripheral nervous systems. NPY has been identified as the most abundant peptide present in the mammalian central nervous system, which consists of the brain and spinal cord.(Heilig & Widerlov, 1995)
Although NPY has many properties to be a potential CNS medication, a fundamental obstacle for the development of NPY as a CNS medication is that it does not readily passively cross the BBB following exogenous administration. The BBB is a highly selective barrier that limits large or hydrophilic molecules from readily entering the brain. This restrictive barrier limits the range of compounds and scaffolds available to medicinal chemistry to develop new chemical entities for the treatment of CNS disorders.
Several strategies have been used to deliver therapeutics across the BBB. An attractive strategy to target drugs across the BBB and into the brain is to use endogenous active transport systems such as receptor-mediated transport (RMT) (Pulicherla & Verma, 2015). RMT is a class of transport system that transports macromolecules such insulin and transferrin across the BBB between the blood and the brain. Transferrin receptors are known to be expressed in the luminal membrane of capillary endothelium of the BBB. For this reason, transferrin (Tf) has been widely studied and shown to be a promising molecular probe for targeted drug delivery to the brain through RMT (Gan & Feng, 2010; Gatter, Brown, Trowbridge, Woolston, & Mason, 1983; Pardridge, 2007). Tf is a single chain 80kDa protein. It facilitates the movement of iron between the blood and brain through RMT. Tf-bound iron from blood binds to the Tf receptor (TfR) expressed on the BBB. Although TfR is expressed on intestinal cells, hepatocytes, and monocytes, they are overexpressed in brain capillary endothelium. Thus Tf-conjugated drug delivery systems (nanoparticles, liposomes and micelles) can improve drug transport across the BBB (Pang et al., 2011; Qian, Li, Sun, & Ho, 2002; Ulbrich, Hekmatara, Herbert, & Kreuter, 2009; Wiley, Webster, Gale, & Davis, 2013; Yan et al., 2013; Ying et al., 2010). Previous studies have extensively examined the Transferrin (Tf) system as a way of targeting drug loaded nanoparticulate formulations to the brain (Broadwell, Baker-Cairns, Friden, Oliver, & Villegas, 1996; Pardridge, 2014; Qian et al., 2002; Ulbrich et al., 2009). Tf can be adsorbed on or conjugated to different polymers including PLGA, polyethyleneglycol, lipopolyplexes, polymer-based cyclodextrin, and gold nanoparticles (Li, Ogris, Wagner, Pelisek, & Ruffer, 2003; Pan et al., 2008; Sahoo & Labhasetwar, 2005; Zhang et al., 2009). Recent studies have also described a new brain targeting ligand called Rabies Virus Glycoprotein (RVG), which putatively binds to nicotinic cholinergic receptors on the BBB to engage brain uptake of drug formulations (Liu et al., 2016). Both Tf and RVG are likely to be effective in the transport of neuropeptides into the brain as they have been used previously to target therapeutic peptides to the brain. However, to the best of our knowledge, no previous studies have examined Tf and RVG in the brain targeting of Neuropeptide Y using nanoparticles.
Nanotechnology such as nanoparticles can be used to encapsulate our therapeutic peptide. These peptide loaded nanoparticles can be coated with Tf or RVG to utilize receptor-mediated transport, more specifically transcytosis to move the therapeutic across the BBB. Previous studies have utilized a variety of approaches for sustained delivery and brain targeting of drug formulations. Gan et al. developed a Tf conjugated nanoparticulate system to deliver clinical Taxotere across the BBB and showed that Tf-conjugated nanoparticles were more effective in reaching the brain compared to unconjugated nanoparticles (Gan & Feng, 2010). Therapeutic peptides can be encapsulated within different polymers including Poly(lactic-co-glycolic acid) (PLGA), Bovine serum albumin (BSA), polyethyleneglycol, lipopolyplexes, polymer-based cyclodextrin, and gold nanoparticles (Li et al., 2003; Pan et al., 2008; Sahoo & Labhasetwar, 2005; Zhang et al., 2009). Smaller nanoparticles (under 200 nm) can potentially increase the penetrance of our formulation through the BBB and into the brain as size plays a role in brain penetrance. We focused our efforts on using PLGA and BSA for our nanoparticle polymers because they are biocompatible, biodegradable, and are approved by the US Food and Drug Administration (FDA) (Makadia & Siegel, 2011). We also have extensive experience and expertise working with these polymers. For example, we have previously formulated PLGA and BSA based Oxytocin loaded nanoparticles that can potentially be targeted to brain (Uz Zaman et al., 2018). We have also used PLGA to develop nanoparticulate dosage forms to deliver the anti-cancer drug camptothecin to B16 cells in vitro (Tong, Wang, & D’Souza, 2003). Additionally, previous studies carried out in our lab in non-human primates have shown low levels of toxicity using BSA as the polymer for particulate drug delivery (Oettinger et al., 2007). Furthermore, both PLGA and BSA nanoparticles have sustained release properties, which can increase the half-life of NPY and thereby decrease the frequency of administration. Both of these will result in increased patient compliance. One of our goals was to find a release profile that provides an initial burst release of the peptide to induce rapid symptom control followed by a sustained release for prolonged therapeutic effects.
In order to increase the brain penetrance and extend the duration of action of NPY, in the present study, we have designed two polymeric-based nanoparticle formulations. Once the optimization of the formulation was done, we have determined the physical, physiochemical, and release properties of the formulations. We have also determined their potential for undesirable immunogenic reactions and cytotoxicity. In order to select a formulation based on our criteria for a desired small particle size and optimal release profile, we compared formulations composed of PLGA and BSA particles conjugated with Tf or RVG. We found that, of the four final formulations, NPY-loaded BSA nanoparticles conjugated to RVG produced the smallest-sized particles and maintained our desired release profile of NPY.
Materials and methods
Materials
Poly(D,L-lactide-co-glycolide) lactide:glycolide (50:50), polyvinyl alcohol, dichloromethane, indocyanine green, transferrin (Tf), and benzalkonium chloride were purchased from Sigma Aldrich (Milwaukee, WI, USA). Bovine serum albumin (Fraction V), glutaraldehyde (25% in water) and acetone were purchased from Fischer Scientific (Pittsburgh, PA, USA). Neuropeptide Y, Rabies Virus Glycoprotein (RVG) was purchased from GenScript (Piscataway, NJ, USA). All the chemicals were of analytical grade and used as is without further modification.
Preparation of nanoparticles
PLGA nanoparticles were prepared following multiple emulsion solvent evaporation method. The nanoparticles suspension was lyophilized to get dry powder. Briefly, 100 mg of PLGA and 20 μl of sorbitan monooleate were dissolved in 5 ml dichloromethane. 10 mg of NPY was dissolved in 1 ml of deionized water. The water/oil emulsion of aqueous NPY and organic PLGA solution was prepared by homogenization at 35,000 rpm for 1 minute. The emulsion was subjected to probe sonication for 5 minutes with a 30 second pause after each minute. This primary emulsion was added drop wise to 10 ml of a 1% w/v polyvinyl alcohol solution and homogenized using the same method as described for primary emulsion in order to obtain the final water/oil/water (w/o/w) emulsion. The emulsion was kept under stirring conditions for 6 hours to remove dichloromethane. Once all dichloromethane was evaporated, trehalose (2% w/v) was added as a cryoprotectant and the formulation was lyophilized. The lyophilized particles were stored in glass vials at -20oC for future use.
Coacervation/ nanoprecipitation method was used to make BSA based nanoparticles. BSA nanoparticles were also lyophilized to get dry powder form. Briefly, 270 mg of BSA and 27 mg of NPY were dissolved in 10 ml of 10mM NaCl at pH 9.3. Acetone was added drop wise at a speed of 1ml/minute to the aqueous solution. Acetone to aqueous solution ratio was 2:1 and the stirring speed was 800 rpm. After desolvation, the nanoparticle suspension was cross-linked for 30 minutes with 25% (w/v) glutaraldehyde in water (200 μL of glutaraldehyde for 1000 mg of BSA). Excess of glutaraldehyde was neutralized with sodium bisulphite. Trehalose (2% w/v) was added as a cryoprotectant and the nanoparticle suspension was lyophilized as above.
Conjugation of brain targeting ligands to peptide-loaded nanoparticles
Tf and RVG have been used as brain targeting ligands in several previous studies (Blesch & Tuszynski, 2007; Gan & Feng, 2010; Kim, Choi, Kim, & Tae, 2013). We attempted to adopt these approaches for the formulation of NPY loaded nanoparticles. Tf and RVG were conjugated to PLGA and BSA nanoparticles in two steps as previously described (Uz Zaman et al., 2018). The first step required activation of the carboxylic acid terminal groups using 1-ethyl-3-(3-dimethylaminopropyl) carbodimide (EDC) and NHS (98%) followed by conjugating Tf or RVG in the nanoparticles. Four lead formulations that were prepared are listed in Table 1.
Particle size, zeta potential and surface morphology
Malvern Zetasizer (Malvern Instruments Inc, Massachusetts, USA) was used to measure the particle size and zeta potential of the nanoparticles. In order to determine the size and zeta potential, a 50 μL aliquot of the nanoparticle suspension was diluted with 1.5 mL distilled water and then measured in Malvern Zetasizer. The results reported are average values from triplicate runs of three independent experiments for each sample. Surface morphology of the nanoparticles was investigated using a scanning electron microscope (Phenom World Pure Scanning electron microscope, AZ, USA). Nanoparticles were mounted onto metal stubs using double-sided adhesive tape and after being vacuum coated with a thin layer (100–150A°) of gold, the nanoparticles were observed under 20kV using Phenom scanning electron microscope.
Peptide encapsulation efficiency
In order to determine the encapsulation efficiency, a known amount of each nanoparticle suspension was centrifuged at a speed of 35,000 rpm. The amount of peptide in the supernatant was determined by an NPY-specific enzyme linked immunosorbent assay (ELISA) (NPY ELISA kit, Catalogue no: EZHNPY-25K, EMD Millipore Corporation, Billerica, MA 01821 USA.). To determine the peptide encapsulation efficiency, this amount was subtracted from the total amount of peptide used for the formulation.
Fourier Transform Infrared spectroscopy (FT-IR) to determine conjugation efficiency
FT-IR spectroscopy was used to determine the conjugation efficiency of Tf and RVG on the surface of PLGA and BSA nanoparticles. The FT-IR spectra were recorded using IRAffinity -1S Spectrometer (Shimadzu Corporation, Japan). FTIR spectra of transferrin and transferrin (Tf) and RVG coated PLGA and BSA nanoparticles were obtained in the range of 4000 to 600 cm-1. Transmittance as a function of wave number was used to record the spectral output.
Immunogenicity
The amount of nitrite released by dendritic cells (DC) in presence of nanoparticulate formulations was used as an indicator of the potential of the formulations to induce an immunogenic reaction. Murine dendritic cells (DC 2.4, Millipore Sigma) were grown in DMEM with glucose, L-glutamine, and 10% FBS and were plated in a 96 well-plate at 15,000 cells/well. The cells were stimulated with 300µg blank nanoparticles (1 mg/ml stock suspension) or peptide nanoparticles (1 mg/ml stock suspension) using incomplete DMEM for 16 hrs (n=3). The treatments consisted of untreated cells (negative control), the 4T1 immunogenic breast cancer vaccine (positive control), blank particles, and peptide particles. After 16 hours of incubation, the supernatant was collected and analyzed for nitric oxide concentration using the Greiss chemical method. The Greiss reagent was prepared by mixing equal volumes of 1% sulfanilamide and 0.1 % N-(1-napthylethyldiamine) solutions. One hundred microliters of the supernatant was transferred to a 96 well plate, to which 100µl of Greiss reagent was added. The plate was incubated for 10 minutes and read at 540 nm to assess the color change using a microplate reader (Synergy H1; BIO-TEK Instruments, Winooski, VT). A standard curve of NaNO2 (1mM stock concentration in distilled water further diluted to the highest standard at 100 µM followed by serial dilutions to 1.56 µM) was used for the calculation of nitrite concentration in each well.
Cytotoxicity
MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to assess the cytotoxicity of blank and peptide loaded nanoparticles. Briefly, Cell suspension equivalent to 10,000 cells/well was plated in a 96-well cell culture plate, and complete DMEM medium was added to obtain a volume of 100 µL per well followed by addition of various concentrations of the nanoparticles ranging from 0.5 µg /mL to 1000 µg/mL in triplicates and incubation for specified time points (24 and 48 hours). Untreated cells (cells only) and benzalkonium chloride were used as negative and positive controls, respectively. In each wells, 10 μL of MTT Reagent was added followed by incubation at 37°C for 2 to 4 hours. After the incubation, when the purple precipitate was clearly visible 100 μL of detergent reagent was added to all wells, including controls. Absorbance in each well was measured at 570 nm in a microtiter plate reader. The formation of the colored product was quantified by measuring the absorbance and was correlated to the percentage of living cells.
In vitro release
The release pattern of NPY from nanoparticles was studied in phosphate buffered saline (PBS, pH 7.4) and in 10% mice serum. Mice serum was collected from Swiss-Webster mice by submandibular bleeding (Golde, Gollobin, & Rodriguez, 2005). Briefly, 10 mg of nanoparticles were suspended in 1 mL of release media, PBS or 10% mice serum. Nanoparticles suspension in release media was kept for continuous shaking (100rpm) at two different temperatures, 4°C and 37°C. At each time point, the tube was centrifuged at 35,000 rpm for 15 min and the supernatant was removed. Sample volume was replenished with media. The supernatant (n = 3) was analyzed for the amount of NPY released in the media using a NPY ELISA as mentioned earlier.
Results
Particle size, zeta potential and surface morphology
Size of the nanoparticles play a pivotal role in active transport across the BBB (Etame, Smith, Chan, & Rutka, 2011; Hanada et al., 2014; Saraiva et al., 2016; Sonavane, Tomoda, & Makino, 2008). The mean diameter of NPY loaded PLGA nanoparticles ranged between 204.9 and 267.6 nm. Charge of the PLGA-based particles ranged from -16.3 to -20.10 mV. The mean diameter of the NPY loaded BSA nanoparticles ranged from 140.3 to 197 nm in diameter. All the BSA nanoparticles were negatively charged ranging from -16.00 to -20.5 mV. The lowest particle size (approx. 137nm) was obtained using a BSA formulation and among NPY loaded nanoparticles RVG conjugated BSA particles were the smallest (140.3nm)
Conjugation of brain targeting ligands to peptide loaded nanoparticles
Figures 2 (A and B) shows the FT-IR spectra of Tf- and RVG-unconjugated and conjugated PLGA nanoparticles. Figure 2 (A) shows the FT-IR spectrum of PLGA nanoparticles conjugated with Tf showing the amide bands assigned to the protein. The peak of the amide I band in the spectrum of PLGA-Tf nanoparticles occurred at 1648 cm-1. The bands of the characteristic functional groups of PLGA nanoparticles are present (1750 cm-1 for C=O stretching and 1000–1300 cm-1 for the ester C-O stretching), with small shifts when compared to the spectrum of unconjugated nanoparticles. The presence of the amide bond confirms the presence of Tf on the Tf-coated PLGA nanoparticles. Figure 2 (B) shows FT-IR spectrum of RVG conjugated to PLGA nanoparticles containing characteristic peaks of both PLGA and RVG.
Figure 2 (C and D) shows the FT-IR spectra of RVG and Tf conjugated BSA nanoparticles. Both spectra shows two main peaks at 1640 cm-1 and 1535 cm-1,respectively the two peaks of amide I and amide II vibrations of Tf and RVG. The same two peaks are visible for BSA as well. Both RVG- and Tf-conjugated BSA nanoparticles shows intensified peaks for amide I and amide II vibrations indicating conjugation of RVG and Tf to BSA.
Encapsulation efficiency
The encapsulation efficiency was determined before the conjugation of the nanoparticles to the brain targeting ligands (Tf and RVG). The amount of NPY in the supernatant was subtracted from the total amount of peptide used for the formulation to determine the peptide encapsulation efficiency inside the nanoparticles. The NPY content of the PLGA nanoparticles was 78 ± 2.5% (w/w) and for BSA nanoparticles it was 71 ± 2.5% (w/w).
Immunogenicity determination
Nitric oxide release is used as an important indicator of innate immune response (Bogdan, Rollinghoff, & Diefenbach, 2000; Zughaier, Shafer, & Stephens, 2005). Figure 3 shows the effects of each formulation of neuropeptide Y on nitric oxide release. None of the blank or NPY-loaded nanoparticles induced significant nitric oxide release. This shows non immunogenicity of the nanoparticle formulations which is desirable. In contrast, 4T1 murine tumor antigen loaded microparticles resulted in a large NO release and high immunogenicity as the control immunogenicity response.
Cell cytotoxicity
The cytotoxic effects of the four lead nanoparticle formulations (as mentioned in Table 1) at given concentrations are depicted in Figure 4. None of the formulations were cytotoxic within the tested concentration range (0.0625–1 mg/mL) after 48 hours. The percent cell viability is expressed relative to negative controls of cells treated with DMEM medium only. Benzalkonium chloride, used as the positive control was cytotoxic, as demonstrated by 21.44% and 20.089% cell viability after 24 and 48 hours, respectively.
In vitro release
The release of NPY from PLGA nanoparticles in PBS was sustained over a period of 23 days (Figure 5). NPY was released from PLGA nanoparticles in a sustained manner and 50% of peptide was released over a period of 14.5 days (Figure 5). The release of NPY from BSA nanoparticles in PBS was sustained over a period of 20 days with 50% of NPY released over a period of 12 days (Figure 5). In 10% serum at pH 7.4 NPY is released from BSA nanoparticles in sustained fashion, with initial burst release of around 20% and 50% of NPY released over a period of 6 days at both 4oC and 37oC. In order to get a clearer view of the burst release, we conducted a release study for 6 hours with sampling interval of 30 minutes. At the end of 6 hours, 31.30% and 30.25% of the NPY has been released from the BSA nanoparticles at 4oC and 37oC temperature, respectively. In contrast, only 9.65% and 8.90% of the NPY has been released from the PLGA nanoparticles at 4oC and 37oC temperature, respectively. (Figure 6). These data show that BSA nanoparticles have a faster initial release profile than PLGA particles, and that temperature does not have a significant effect on the release profile over the first 6 hours. Our goal was to find a release profile that provides an initial burst release of the peptide to induce rapid symptom control followed by a sustained release for prolonged therapeutic effects. Figure 7 demonstrates that the RVG-conjugated BSA nanoparticles have a mixed burst release / sustained release profile, with approximately 38% release within the first 6 hours, but 50% release not occurring until approximately 6 days in solution.
Discussion
The aim of this study was to formulate several NPY loaded nanoparticulate formulations that can potentially cross the BBB. All 4 of the nanoparticle formulations were extensively characterized for their physiochemical properties and release profiles after they were produced. We used two polymers, PLGA and BSA for formulating nanoparticles because they are biocompatible, biodegradable and approved by the US Food and Drug Administration (FDA) (Makadia & Siegel, 2011). Additionally, we have past experience working with these polymers in a formulation to delivery of a therapeutic peptide across the BBB. We focused to achieve brain targeting of our nanoparticles by conjugating the nanoparticles to either Tf or RVG, which are well documented brain targeting ligands. We incorporated the proposed polymers and brain targeting ligands to produce four lead formulations each containing one brain targeting ligand, one polymer for the particle matrix and the neuropeptide Y, the therapeutic agent. Thus we got four lead formulations: Tf-PLGA-NPY, Tf-BSA-NPY, RVG-PLGA-NPY, and RVG-BSA-NPY. Once we formulated the nanoparticles, we characterized them based on their properties. We selected size and release profile as two criteria to narrow down the optimal formulation. For size, the smaller the particle produced, the better its chance of being transported across the BBB. From past studies, we determined that the particles should be under 200 nm in size. Secondly, we aimed for a release profile, which consisted of initial burst release of the peptide from the formulation followed by a extended sustained release of the peptide over time. From previous studies we believe that this would the optimal profile for therapeutic effects.
Delivering therapeutics across the BBB to the brain is a major obstacle for the treatment of many CNS disorders. Receptor-mediated transport (RMT) has been described in many studies as an appealing strategy for delivering drugs to the brain (Pulicherla & Verma, 2015). Transferrin has been widely studied as a brain targeting ligand. Tf has shown to be a promising molecular probe for targeted drug delivery to the brain through RMT as transferrin receptors are known to be expressed in the luminal membrane of capillary endothelium of the BBB (Gan & Feng, 2010; Gatter et al., 1983; Pardridge, 2015). Recently several studies have descried Rabies virus glycoprotein (RVG) as a promising brain targeting ligand. RVG is a short peptide that can bind to nicotinic cholinergic receptors on the BBB. It has been employed as a targeting ligand to deliver small interfering RNA (siRNA) to the brain (Kumar et al., 2007; Son et al., 2011). RVG tagged PEGylated cyclodextrin (CD)-based nanoparticle has been used in another study to deliver siRNA to the brain [50]. The BBB is one of the most difficult biological barriers to cross in order to deliver therapeutic agents/ drugs/ peptides for treatment of neurological diseases [28, 51, 52]. But these promising studies have created enthusiasm that feasible approaches are possible to deliver therapeutics across the BBB to the brain using RMT. If a reliable vehicle system for active transport into the brain could be developed, that would allow for a broad range of compounds that have poor passive brain bioavailability to readily penetrate the brain, it would be a paradigm shifting advance for CNS drug discovery. There are hundreds to thousands of compounds with potentially therapeutic use for CNS disorders that have been abandoned because of poor brain bioavailability. Some of these compounds include neurotrophins such as BDNF [9, 10], secretase inhibitors for Alzheimer’s disease [11], anti-inflammatory cytokines [12, 13], and therapeutic plasmid DNA for neuronal stimulation or myelin recovery and remodeling [14-16]. Neuropeptides such as neuropeptide Y, BDNF, orexin, and others are have defined disease states where they show profound therapeutic promise, including epilepsy, substance-use disorders, and obesity.
The size, shape, and charge of our particles are highly critical for delivering drugs across the BBB [53]. Nanoparticles having sizes smaller than 500nm are usually taken via the clathrin-mediated transport system whereas larger particles are usually transported via macropinocytosis [54]. In reference to charge, negatively-charged nanoparticles are endocytosed by interacting with the positive site of the proteins on membrane. It has been reported in a previous study that the negatively charged nanoparticles are highly endocytosed by cells due to their repulsive interactions with the negatively charged cell surface [54, 55], and positively charged particles show substantially reduced BBB penetrance. Our nanoparticle formulations exhibit a smooth, regular and spherical shape, which helps maximize uptake, as spherical particles are efficiently taken up via both clathrin and caveolae mediated transport [56, 57]. It is desirable to have particles below 200 nm as this will maximize the probability of efficient brain uptake. In initial studies, we generate PLGA particles conjugated to Tf using homogenization. However, the particles size exceeded 200 nm with this approach. We modified the production method to sonication and were able to readily achieve particles sizes below 200 nm. Such sizes were also readily achievable with BSA nanoparticles. However, it is important to note the BSA particles conjugated to RVG clearly achieved the smallest particles sizes.
In order to demonstrate the therapeutic viability of our nanoparticulate formulations, we have completed both immunogenicity and cytotoxicity studies in vitro. As confirmed in our past research, our nanoparticle formulations have shown to be both safe and non-toxic in non-human primates. In primates, all parameters related to liver toxicity (such as SGOT, SGPT) and kidney toxicity (such as serum creatinine, serum bilirubin) were normal after repeated nanoparticulate administrations when compared to the control group. Also, no antibody titers in response to our polymeric particle matrix were detected demonstrating the absence of an immune response to the polymer matrix formulation [36]. For confirmation of a potential innate inflammatory response, nitric oxide assays are an important tool. For the assay, dendritic cells are plated in a well plate and exposed to the formulation. After 16 hours, using reagents, a stronger color change indicates a higher production of nitric oxide by these antigen-presenting cells in response to our formulation’s antigenicity. We have demonstrated that there is no significant difference in the amount of nitric oxide released in the supernatant of cells exposed to peptide-loaded nanoparticles compared to blank nanoparticles or the cells alone. This indicates minimal if not negligible immunogenicity of our formulation. For reference, nitric oxide release in response to our OT-loaded particles was substantially lower compared to cells that were exposed to a well-known highly-immunogenic breast cancer vaccine formulation. Likewise, cytotoxicity of blank nanoparticles was assessed by MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay in a macrophage (RAW 246.7) cell line. We found no significicant celll death following 48 hours of exposure to our formulations indicating that our formulation was not significantly toxic to the cells.
A major advantage of a nanoparticle formulation for peptide delivery is that the encapsulation protects the peptide from degradation, and sustains the delivery of the therapeutic agent over an extended period of time. This sustained delivery allows for fewer administrations and easier compliance, an issue of particular importance in the treatment substance abuse disorders for a prolonged amount of time or in cases where patients have difficulty with compliance. Resultantly, the NPY can provide long-lasting therapeutic affects in the brain, improving the health of those suffering with various cognitive ailments. We have demonstrated that our formulations maintain a sustained release profile of NPY in vitro in both PBS and plasma, which was confirmed using ELISA (Figure 5 and 7). During our release studies, we took samples in 30-minute interval initially, then hourly, and then daily to establish a release profile covering several weeks. In order to select the most viable formulation to test in vivo, we explored the two factors discussed prior 1) size and 2) suitable sustained release criteria. Our results showed that the RVG-conjugated BSA nanoparticles were smaller in size compared to the remaining three nanoformulations. For the purpose of our treatment with NPY, a desired release profile consists of an initial burst release followed by a sustained release for several days. The NPY released in 6 hours in 37 degrees Celsius serum was as follows for both particulate matrices: BSA nanoparticles (30.25% release) and PLGA nanoparticles (8.9% release). Given the release profile of PLGA nanoparticle formulations, they would not be viable candidates since they released NPY too slowly to produce a significant therapeutic window for NPY in the brain. Based on the desired release profile of NPY from BSA nanoparticles, we concluded that Tf-conjugated BSA particles RVG-conjugated BSA particles would be ideal for the preferred release parameters. Therefore, when selecting the final formulation to test in vivo, we determined that our BSA nanoparticles conjugated to RVG formulation was the most suitable candidate. Because they were smaller than the size of Tf-conjugated BSA microparticles, which could aid in enhanced penetrance across the BBB. Additionally, they demonstrated the desired initial and sustained release profile of NPY in the brain and could be utilized as the lead formulation to test in an in vivo murine model.
Conclusion
In this study, we have successfully formulated Tf or RVG-conjugated biodegradable PLGA and BSA nanoparticles loaded with oxytocin. We have used solvent evaporation and nanoprecipitation/coacervation methods for formulating PLGA and BSA nanoparticles respectively. Standard in vitro characterization of the nanoparticles using various methods such as zeta sizing and scanning electron microscopy has been done. All four nanoparticulate formulations were uniformly distributed in nm range, spherically shaped, and individually dispersed. Standard assays proved that all four formulations were non-immunogenic and non-toxic. In case of release pattern, all of them showed sustained delivery of NPY allowing for fewer administrations and easier compliance. All these findings strongly suggest that our nanoparticulate formulation could serve as a potential candidate for delivering NPY across the BBB non-invasively. Among the four formulations, the NPY-loaded BSA nanoparticles conjugated to RVG formulation produced particles that were smallest in size and demonstrated a desirable release profile. The smallest size of this formulation make it a very strong candidate for the delivery of NPY to the brain. We further propose testing our selected NPY loaded nanoparticle formulation in vivo in well-characterized murine disease models to determine the efficacy of this delivery method
Acknowledgements
The authors acknowledge the use of facilities of Vaccine Nanotechnology Laboratory, Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, Mercer University, Atlanta, GA. The results of the study will be a part of Rokon Uz Zaman’s dissertation.
Funding
These studies were supported by the National Institutes of Health [NS100512 (KSM and MJD)] and by funding from the Mercer University College of Pharmacy.
Disclosure
The authors report no conflicts of interest in this work. The authors alone are responsible for the content and writing of this article.
Tables:
Table 1: PLGA and BSA nanoparticle formulations
| Number | Method of preparation | Formulation |
| 1 | Sonication/Solvent evaporation | TF conjugated to PLGA nanoparticles |
| 2 | Sonication/Solvent evaporation | RVG conjugated to PLGA nanoparticles |
| 3 | Coacervation/nanoprecipitation | TF conjugated to BSA nanoparticles |
| 4 | Coacervation/nanoprecipitation | RVG conjugated to BSA nanoparticles |
This table shows the four lead formulations developed in the present study. The abbreviations include Tf = Transferrin; RVG = Rabies Virus Glycoprotein; PLGA = poly(lactic-co-glycolic acid); BSA = Bovine serum albumin;
Table 2: Size distribution and Zeta potentials of different PLGA and BSA nanoparticle formulations of neuropeptide Y.
| Formulation | Characteristics | ||||||
| Base Material | Loading | Targeting Ligand | Preparation Method | Size (nm) | Standard deviation | Zeta Potential (mV) | Standard Deviation |
| PLGA | Blank | Tf | Homogenization | 240 | 0.72111 | -20.10 | 0.5131 |
| PLGA | NPY | Tf | Homogenization | 267.6 | 3.50235 | -19.5 | 0.3754 |
| PLGA | Blank | Tf | Sonication | 205.2 | 0.45825 | -16.3 | 0.6244 |
| PLGA | NPY | Tf | Sonication | 204.9 | 1.54138 | -17.4 | 0.3427 |
| PLGA | Blank | RVG | Sonication | 203.44 | 0.94155 | -16.5 | 0.7094 |
| PLGA | NPY | RVG | Sonication | 205.8 | 3.34713 | -16.5 | 0.8134 |
| BSA | Blank | Tf | Coacervation/
nanoprecipitation |
192.9 | 2.68886 | -19.6 | 0.5 |
| BSA | NPY | Tf | Coacervation/
nanoprecipitation |
197 | 5.03318 | -20.2 | 0.4885 |
| BSA | Blank | RVG | Coacervation/
nanoprecipitation |
137 | 3.67151 | -20.5 | 0.4932 |
| BSA | NPY | RVG | Coacervation/
nanoprecipitation |
140.3 | 0.45310 | -16.0 | 0.4831 |
Abbreviations include, Tf = Transferrin; RVG = Rabies Virus Glycoprotein; PLGA = poly(lactic-co-glycolic acid); BSA = Bovine serum albumin; NPY = Neuropeptide Y
Figures and legends:
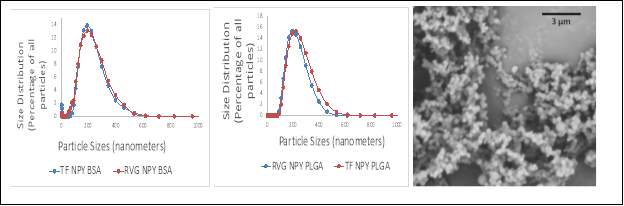
Figure 1. Representative particle sizes and morphology of our nanoparticle formulations. (Left) Size distribution of PLGA nanoparticles conjugated to Tf or RVG, (Middle) size distribution of BSA nanoparticles conjugated to Tf or RVG, and (Right) scanning electron microscopy images of the BSA nanoparticles conjugated to Tf at a 3um resolution. The BSA nanoparticles conjugated to RVG were too small to image at a 3um resolution.
Tf = Transferrin; RVG = Rabies Virus Glycoprotein; PLGA = poly(lactic-co-glycolic acid); BSA = Bovine serum albumin; NPY = Neuropeptide Y
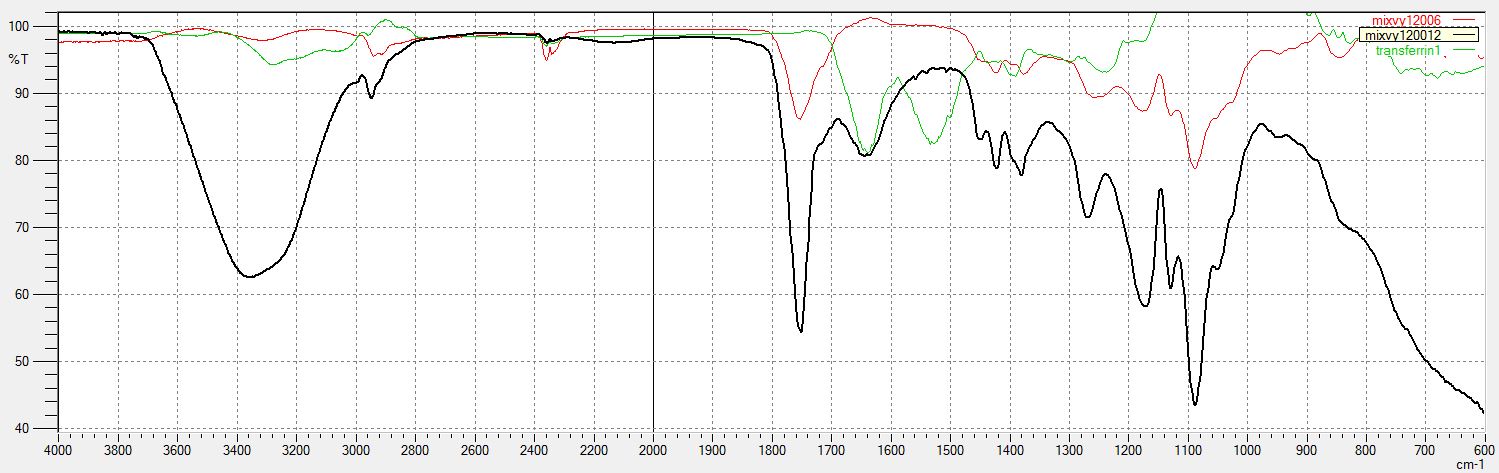
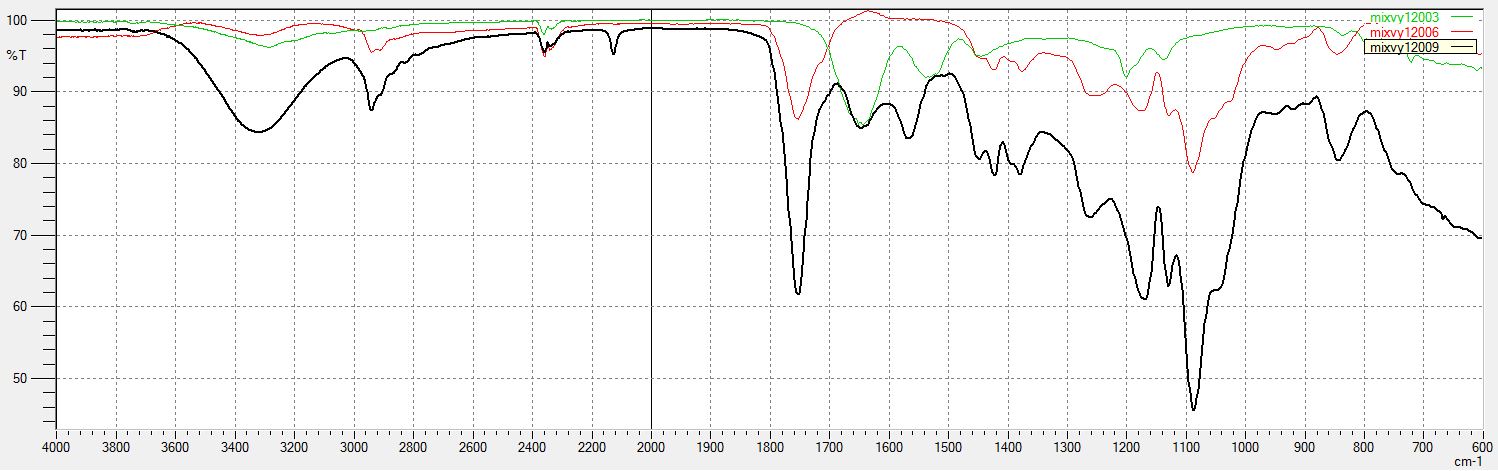
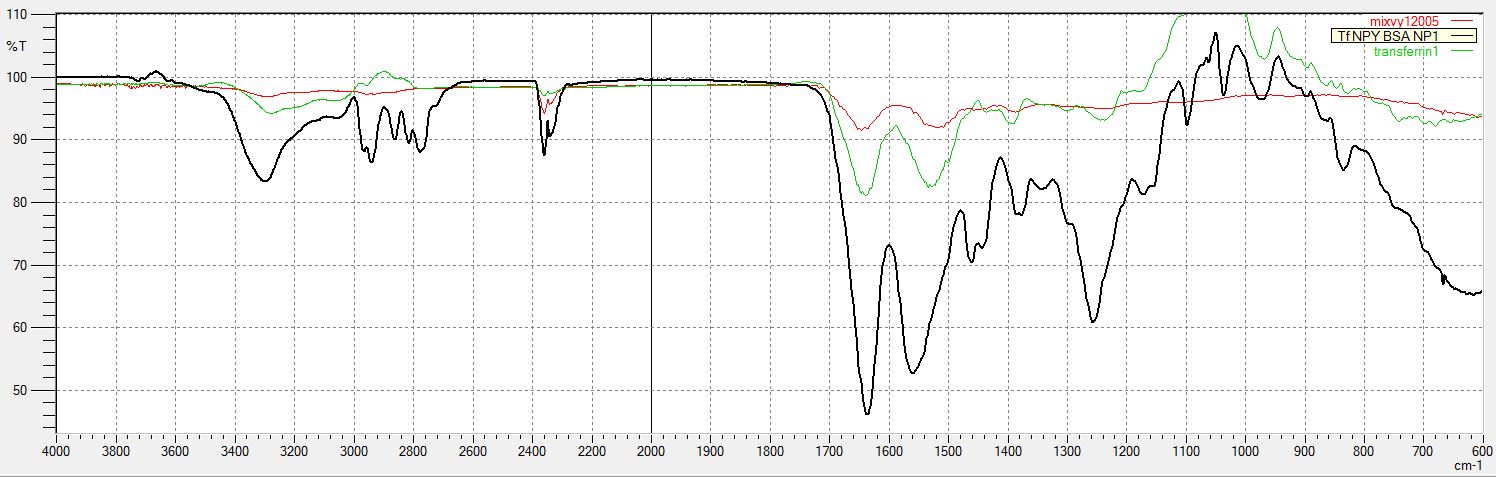
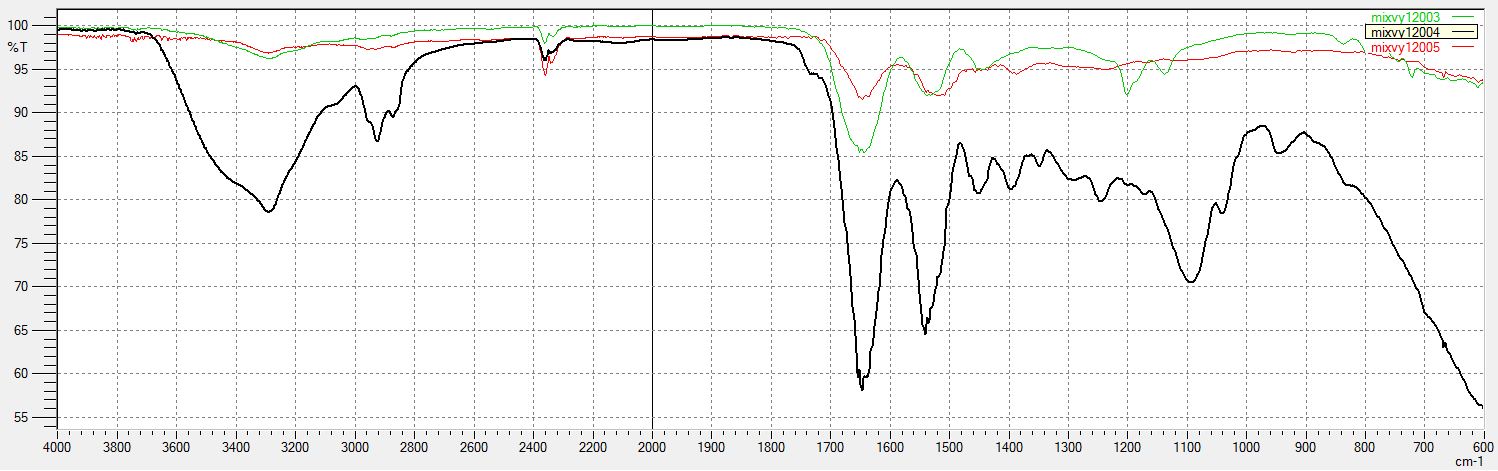
Figure 2. FT-IR spectrum data collected to confirm the conjugation of the brain targeting ligands to the nanoparticles. (A): FT-IR spectrum of Tf conjugated to PLGA nanoparticles. In the spectra of Tf shows two main peaks at 1648 cm-1 and 1550 cm-1 that can be assigned to the amide I and amide II vibrations, respectively. FT-IR spectrum of blank and unconjugated PLGA nanoparticles shows characteristic bands of carbonyl C=O stretching (1750 cm-1) and the ester C-O stretching (1000–1300 cm-1). FT-IR spectrum of PLGA nanoparticles conjugated to Tf shows the amide bands assigned to the protein. The peak of the amide I band in the spectrum of PLGA-Tf nanoparticles occurs at 1648 cm-1. The bands of the characteristic functional groups of PLGA nanoparticles are present (1750 cm-1 and 1000–1300 cm-1) with small shifts when compared to the spectrum of the unmodified nanoparticles. The presence of amide I band in the PLGA-Tf spectrum demonstrates that Tf has been conjugated to the PLGA nanoparticles. (B): FT-IR spectrum of RVG conjugated to PLGA nanoparticles. (C): FT-IR spectrum of Tf conjugated to BSA nanoparticles. (D): FT-IR spectrum of RVG conjugated to BSA nanoparticles. Both BSA spectra show two main peaks near 1650 cm-1 and 1540 cm-1, which are the two peaks of the amide I and amide II vibrations of Tf and RVG, indicating conjugation of RVG and Tf to BSA.
Tf = Transferrin; RVG = Rabies Virus Glycoprotein; PLGA = poly(lactic-co-glycolic acid); BSA = Bovine serum albumin; NPY = Neuropeptide Y; FT-IR = Fourier Transform Infrared spectroscopy
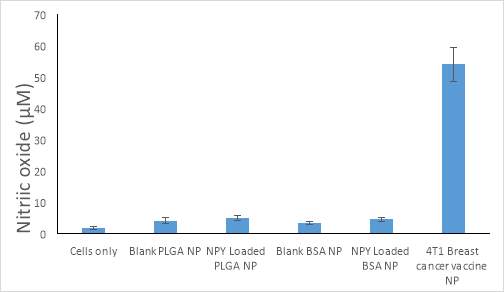
Figure 3. Induction of nitric oxide release by our lead neuropeptide Y formulations. The neuropeptide Y formulation was generated by double emulsion solvent evaporation by sonication. All points represent the mean ± standard error. Abscissae: Various formulations compared to the negative control of untreated cells and the positive control of the 4T1 breast cancer vaccine that induces a robust immune response. Ordinates: Measured concentration of nitric oxide expressed on an absolute scale in units of micromolar. All experiments were conducted in triplicate.
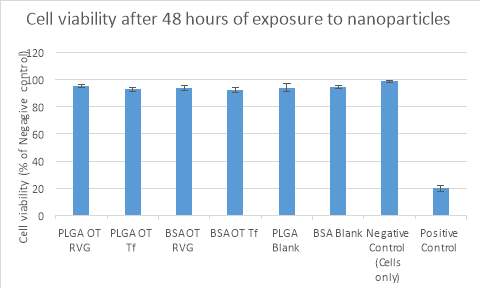
Figure 4. Cytotoxicity profile of the nanoparticle formulations. After 48 hours of exposure, none of the four formulations were cytotoxic across the tested concentration range (0.0625–1 mg/mL). As there were no differences between the concentrations tested, only the highest concentration (1 mg/mL) is shown. Abscissae: Various formulations compared to the negative control of untreated cells and the positive control of benzalkonium chloride (a cationic surfactant that has biocidal activity). Ordinates: Cell viability expressed as a percentage of the negative control at 6 hours. All experiments were conducted in triplicate.
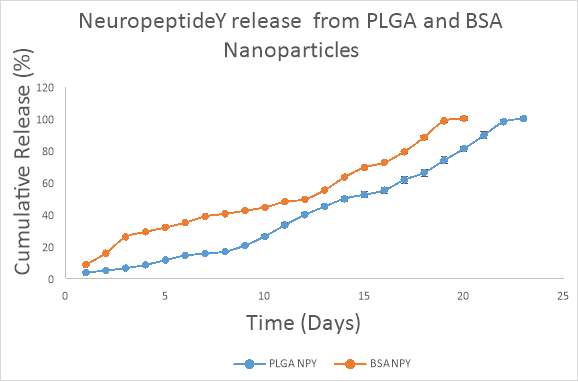
Figure 5. Release of NPY from PLGA and BSA nanoparticles in PBS, pH 7.4. The release of NPY from PLGA nanoparticles was sustained over a period of 23 days. NPY was released from PLGA nanoparticles in sustained fashion with 50% of the NPY released over a period of 14.5 days. The release of NPY from BSA nanoparticles was sustained over a period of 20 days and 50% of OT was released over a period of 12 days. Abscissae: Day following addition of the formulation to solution. Ordinates: Measured concentration of free neuropeptide Y in solution expressed as a percentage of the maximally measured release.
PLGA = poly(lactic-co-glycolic acid); BSA = Bovine serum albumin; NPY = Neuropeptide Y
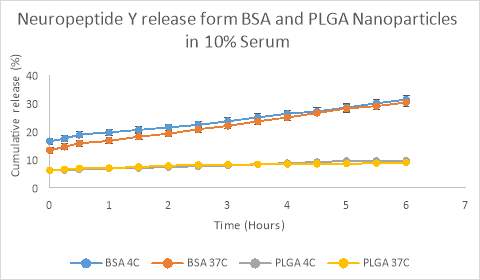
| 31.2954 | 30.25252 | 9.652033 | 8.895914 |
Figure 6. Release OT from PLGA (prepared by sonication) and BSA nanoparticles in 10% serum, pH 7.4 over 6 hours at 4oC and 37oC. At the end of 6 hours, 31.30% and 30.25% of the NPY has been released from the BSA nanoparticles at 4oC and 37oC temperature, respectively. In contrast, only 9.65% and 8.90% of the NPY has been released from the PLGA nanoparticles at 4oC and 37oC temperature, respectively. Abscissae: Hour following addition of the formulation to solution. Ordinates: Measured concentration of free neuropeptide Y in solution expressed as a percentage of the maximally measured release.
PLGA = poly(lactic-co-glycolic acid); BSA = Bovine serum albumin; NPY = Neuropeptide Y;
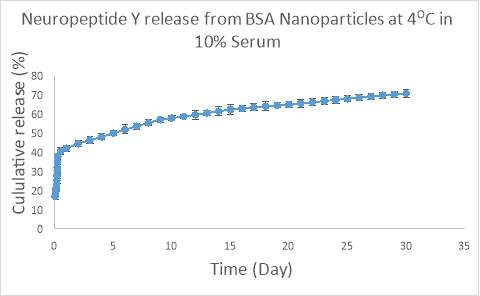
Figure 7. Release NPY from BSA nanoparticles in 10% serum, pH 7.4 at 4oC. 50% of the NPY was released over a period of 6 days. Abscissae: Day following addition of the formulation to solution. Ordinates: Measured concentration of free NPY in solution expressed as a percentage of the maximally measured release.
BSA = Bovine serum albumin; NPY = Neuropeptide Y;
References
Blesch, A., & Tuszynski, M. H. (2007). Transient growth factor delivery sustains regenerated axons after spinal cord injury. J Neurosci, 27(39), 10535-10545. doi: 10.1523/jneurosci.1903-07.2007
Bogdan, C., Rollinghoff, M., & Diefenbach, A. (2000). The role of nitric oxide in innate immunity. Immunol Rev, 173, 17-26.
Broadwell, R. D., Baker-Cairns, B. J., Friden, P. M., Oliver, C., & Villegas, J. C. (1996). Transcytosis of protein through the mammalian cerebral epithelium and endothelium. III. Receptor-mediated transcytosis through the blood-brain barrier of blood-borne transferrin and antibody against the transferrin receptor. Exp Neurol, 142(1), 47-65. doi: 10.1006/exnr.1996.0178
Broderick, D. F., Schweitzer, K. J., & Wszolek, Z. K. (2009). Vascular risk factors and dementia: how to move forward? Neurology, 73(22), 1934-1935. doi: 10.1212/WNL.0b013e3181bd6a46
Duarte-Neves, J., Pereira de Almeida, L., & Cavadas, C. (2016). Neuropeptide Y (NPY) as a therapeutic target for neurodegenerative diseases. Neurobiol Dis, 95, 210-224. doi: 10.1016/j.nbd.2016.07.022
Etame, A. B., Smith, C. A., Chan, W. C., & Rutka, J. T. (2011). Design and potential application of PEGylated gold nanoparticles with size-dependent permeation through brain microvasculature. Nanomedicine, 7(6), 992-1000. doi: 10.1016/j.nano.2011.04.004
Gan, C. W., & Feng, S. S. (2010). Transferrin-conjugated nanoparticles of poly(lactide)-D-alpha-tocopheryl polyethylene glycol succinate diblock copolymer for targeted drug delivery across the blood-brain barrier. Biomaterials, 31(30), 7748-7757. doi: 10.1016/j.biomaterials.2010.06.053
Gatter, K. C., Brown, G., Trowbridge, I. S., Woolston, R. E., & Mason, D. Y. (1983). Transferrin receptors in human tissues: their distribution and possible clinical relevance. J Clin Pathol, 36(5), 539-545.
Golde, W. T., Gollobin, P., & Rodriguez, L. L. (2005). A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim (NY), 34(9), 39-43. doi: 10.1038/laban1005-39
Hanada, S., Fujioka, K., Inoue, Y., Kanaya, F., Manome, Y., & Yamamoto, K. (2014). Cell-based in vitro blood-brain barrier model can rapidly evaluate nanoparticles’ brain permeability in association with particle size and surface modification. Int J Mol Sci, 15(2), 1812-1825. doi: 10.3390/ijms15021812
Heilig, M., & Widerlov, E. (1995). Neurobiology and clinical aspects of neuropeptide Y. Crit Rev Neurobiol, 9(2-3), 115-136.
Herrmann, N., Chau, S. A., Kircanski, I., & Lanctot, K. L. (2011). Current and emerging drug treatment options for Alzheimer’s disease: a systematic review. Drugs, 71(15), 2031-2065. doi: 10.2165/11595870-000000000-00000
Kim, J. Y., Choi, W. I., Kim, Y. H., & Tae, G. (2013). Brain-targeted delivery of protein using chitosan- and RVG peptide-conjugated, pluronic-based nano-carrier. Biomaterials, 34(4), 1170-1178. doi: 10.1016/j.biomaterials.2012.09.047
Kumar, P., Wu, H., McBride, J. L., Jung, K. E., Kim, M. H., Davidson, B. L., . . . Manjunath, N. (2007). Transvascular delivery of small interfering RNA to the central nervous system. Nature, 448(7149), 39-43. doi: 10.1038/nature05901
Li, Y., Ogris, M., Wagner, E., Pelisek, J., & Ruffer, M. (2003). Nanoparticles bearing polyethyleneglycol-coupled transferrin as gene carriers: preparation and in vitro evaluation. Int J Pharm, 259(1-2), 93-101.
Liu, Y., An, S., Li, J., Kuang, Y., He, X., Guo, Y., . . . Jiang, C. (2016). Brain-targeted co-delivery of therapeutic gene and peptide by multifunctional nanoparticles in Alzheimer’s disease mice. Biomaterials, 80, 33-45. doi: 10.1016/j.biomaterials.2015.11.060
Makadia, H. K., & Siegel, S. J. (2011). Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel), 3(3), 1377-1397. doi: 10.3390/polym3031377
Misra, A., Ganesh, S., Shahiwala, A., & Shah, S. P. (2003). Drug delivery to the central nervous system: a review. J Pharm Pharm Sci, 6(2), 252-273.
Oettinger, C. W., D’Souza M, J., Akhavein, N., Peer, G. T., Taylor, F. B., & Kinasewitz, G. T. (2007). Pro-inflammatory cytokine inhibition in the primate using microencapsulated antisense oligomers to NF-kappaB. J Microencapsul, 24(4), 337-348. doi: 10.1080/02652040601162525
Pan, X., Guan, J., Yoo, J. W., Epstein, A. J., Lee, L. J., & Lee, R. J. (2008). Cationic lipid-coated magnetic nanoparticles associated with transferrin for gene delivery. Int J Pharm, 358(1-2), 263-270. doi: 10.1016/j.ijpharm.2008.02.020
Pang, Z., Gao, H., Yu, Y., Guo, L., Chen, J., Pan, S., . . . Jiang, X. (2011). Enhanced intracellular delivery and chemotherapy for glioma rats by transferrin-conjugated biodegradable polymersomes loaded with doxorubicin. Bioconjug Chem, 22(6), 1171-1180. doi: 10.1021/bc200062q
Pardridge, W. M. (2007). Blood-brain barrier delivery. Drug Discov Today, 12(1-2), 54-61. doi: 10.1016/j.drudis.2006.10.013
Pardridge, W. M. (2014). Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert Opin Drug Deliv, 1-16. doi: 10.1517/17425247.2014.952627
Pardridge, W. M. (2015). Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert Opin Drug Deliv, 12(2), 207-222. doi: 10.1517/17425247.2014.952627
Pulicherla, K. K., & Verma, M. K. (2015). Targeting therapeutics across the blood brain barrier (BBB), prerequisite towards thrombolytic therapy for cerebrovascular disorders-an overview and advancements. AAPS PharmSciTech, 16(2), 223-233. doi: 10.1208/s12249-015-0287-z
Qian, Z. M., Li, H., Sun, H., & Ho, K. (2002). Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev, 54(4), 561-587.
Sahoo, S. K., & Labhasetwar, V. (2005). Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharm, 2(5), 373-383. doi: 10.1021/mp050032z
Saraiva, C., Praca, C., Ferreira, R., Santos, T., Ferreira, L., & Bernardino, L. (2016). Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J Control Release, 235, 34-47. doi: 10.1016/j.jconrel.2016.05.044
Son, S., Hwang, D. W., Singha, K., Jeong, J. H., Park, T. G., Lee, D. S., & Kim, W. J. (2011). RVG peptide tethered bioreducible polyethylenimine for gene delivery to brain. J Control Release, 155(1), 18-25. doi: 10.1016/j.jconrel.2010.08.011
Sonavane, G., Tomoda, K., & Makino, K. (2008). Biodistribution of colloidal gold nanoparticles after intravenous administration: effect of particle size. Colloids Surf B Biointerfaces, 66(2), 274-280. doi: 10.1016/j.colsurfb.2008.07.004
Strecker, K., & Schwarz, J. (2008). Parkinson’s disease: emerging pharmacotherapy. Expert Opin Emerg Drugs, 13(4), 573-591. doi: 10.1517/14728210802596906
Tong, W., Wang, L., & D’Souza, M. J. (2003). Evaluation of PLGA microspheres as delivery system for antitumor agent-camptothecin. Drug Dev Ind Pharm, 29(7), 745-756. doi: 10.1081/ddc-120021774
Ulbrich, K., Hekmatara, T., Herbert, E., & Kreuter, J. (2009). Transferrin- and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood-brain barrier (BBB). Eur J Pharm Biopharm, 71(2), 251-256. doi: 10.1016/j.ejpb.2008.08.021
Uz Zaman, R., Mulla, N. S., Gomes, K. B., D’Souza, C., Murnane, K. S., & D’Souza, M. J. (2018). Nanoparticle formulations that allow for sustained delivery and brain targeting of the neuropeptide oxytocin. Int J Pharm. doi: 10.1016/j.ijpharm.2018.07.043
Wiley, D. T., Webster, P., Gale, A., & Davis, M. E. (2013). Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc Natl Acad Sci U S A, 110(21), 8662-8667. doi: 10.1073/pnas.1307152110
Yan, F., Wang, Y., He, S., Ku, S., Gu, W., & Ye, L. (2013). Transferrin-conjugated, fluorescein-loaded magnetic nanoparticles for targeted delivery across the blood-brain barrier. J Mater Sci Mater Med, 24(10), 2371-2379. doi: 10.1007/s10856-013-4993-3
Ying, X., Wen, H., Lu, W. L., Du, J., Guo, J., Tian, W., . . . Zhang, Q. (2010). Dual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animals. J Control Release, 141(2), 183-192. doi: 10.1016/j.jconrel.2009.09.020
Zhang, X., Koh, C. G., Yu, B., Liu, S., Piao, L., Marcucci, G., . . . Lee, L. J. (2009). Transferrin receptor targeted lipopolyplexes for delivery of antisense oligonucleotide g3139 in a murine k562 xenograft model. Pharm Res, 26(6), 1516-1524. doi: 10.1007/s11095-009-9864-8
Zughaier, S. M., Shafer, W. M., & Stephens, D. S. (2005). Antimicrobial peptides and endotoxin inhibit cytokine and nitric oxide release but amplify respiratory burst response in human and murine macrophages. Cell Microbiol, 7(9), 1251-1262. doi: 10.1111/j.1462-5822.2005.00549.x
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Neurology"
Neurology is the specialist branch of medicine that deals with the treatment of disorders of the nervous system. This means that neurologists concern themselves with issues affecting the brain, the nerves, and the spinal cord.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: