C1q Classical Complement Pathway Inhibition of Alzheimer’s Disease Pathogenesis
Info: 9880 words (40 pages) Dissertation
Published: 16th Dec 2019
1C1q Classical complement pathway inhibition: A novel strategy for the inhibition of Alzheimer’s disease pathogenesis
INTRODUCTION
1.1 Alzheimer’s disease
From the development of the Smallpox vaccine, to the discovery of Penicillin in 1928, scientific research has led to the effective diagnosis, treatment and cure of diseases that previously killed millions. Due to numerous medical and technologica advancements, the average global life expectancy has increased by approximately 23 years in the period between 1960 and 2015 (UN World Population prospects, 2008). With the ever-increasing global population, and an increased average life -span for each individual compared with a 100 years ago; comes the advent of age-related diseases that would previously not be evident in the global population. One such age-related phenomenon is Dementia.
Dementia is a broad categorisation defining neurodegenerative diseases of which the principal observable symptom is severe memory loss. It can develop as a result of global brain insults such as brain injury, brain tumours, normal pressure hydrocephalus, chronic alcohol abuse, vitamin B12 deficiency; or as a secondary effect of other medical conditions such as Huntington’s disease, Multiple sclerosis, HIV/AIDS, Lyme disease, Parkinson’s disease, Pick’s disease and Progressive supranuclear palsy. The cause of dementia determines whether the disease is static (non-progressive) or degenerative (progressive). However, most types of dementia such as Alzheimer’s disease (AD), vascular dementia, dementia with Lewy bodies and frontotemporal dementia are degenerative.
1.2 History of Alzheimer’s Disease
The psychiatrist and neuroscientist Alois Alzheimer first outlined the behavioural and histological hallmarks of this eponymous disorder. Alzheimer recorded the ante-mortem history of his 55 year old patient Auguste Deter who presented with the behavioural symptoms of impaired memory, aphasia, social incompetence, disorientation and delusion (Ramirez-Bermudez 2012). Deter’s cognitive functions further deteriorated with age. After her death in 1906, Alzheimer conducted post-mortem studies in order to identify the neuropathological causes of the patient’s behavioural deficits. He identified what we now describe as amyloid β (Aβ) plaques and neurofibrillary tangles (NFT) in her brain and presented these findings later that year and termed the disease presenile dementia. (Zilka, Novak 2006).
1.3 Epidemiology
AD accounts for 60-70% of all dementia cases. Globally an estimated 35.6 million people were living with dementia in 2010, a number that is set to quadruple to 115.4 million by 2050 unless preventative, therapeutic strategies are developed (Brookmeyer et al., 2007). Most cases of AD occur sporadically i.e., there is no identifiable cause. However, age is the most significant risk factor for sporadic AD (Fig. 1.1). AD affects approximately 5% of people over the age of 65yrs. The prevalence of AD rises exponentially as the patient’s age advances, increasing to approximately 35% in individuals over the age of 85 (Brookmeyer et al., 2002, Kamer et al., 2008, Reitz et al., 2011). Two out of every three AD sufferers are female, though the role Gender plays is as yet unidentified (Alzheimer’s Disease International).
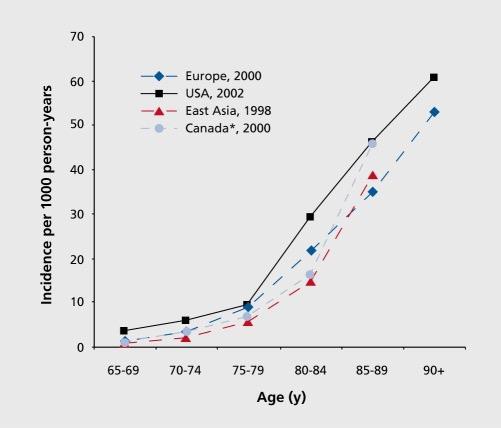

It is currently impossible to ascertain whether the incidence reaches a zenith or continues to rise after 85 years of age (Qiu et al., 2009). This question will likely become more important in future if faced with increased longevity in life span. It may enable researchers to further elucidate whether AD is a disease of the “when” i.e. a disease that will eventually become prevalent in a majority of the aged population if the average life span continues to increase; or whether it is a disease of the proportion who will develop the disorder and the proportion that will not.
There is a substantial socioeconomic and financial cost of caring for patients with dementia, and it is thought to amount to £23 Billion a year in the UK alone, and £380 billion worldwide (Mashta, 2007, Mangialasche et al, 2012). Consequently, dementia and its most common form AD hold the potential of an impending global health crisis that is also too costly economically to ignore.
1.4 Risk factors
For a disease as extensively researched as AD, a thorough understanding of a single causative mechanism underlying the disorder has proved elusive. This lack of clarity is due to the interplay between a number of environmental and genetic risk factors that suggest an increased propensity towards AD and contribute towards this disorder of multifactorial causality. Population-based studies have been the primary approach used in the identification of influential risk factors. As previously stated age confers the most risk for the development of AD. Another major risk factor is genetics. About 5% of all AD suffers are genetically predisposed to the disorder. The onset of symptoms for this familial AD often occurs much earlier than sporadic i.e between 30-65 years (Bird, 2008). Other risk factors for the development of AD include systemic disease, oxidative stress, obesity, reduced physical activity, cerebral hypoperfusion, inflammation, hypoxia, cigarette smoking, excess alcohol consumption, cardiovascular disease, traumatic brain injury, late-life metabolic syndrome and depression (Xu et al., 2013).
1.5 Pathology
Over the past century, post-mortem studies have been the primary method by which to confirm AD diagnosis. The primary pathological hallmarks are deposition of the diffuse and neuritic extracellular beta-amyloid (Aβ)/senile plaques in the parenchyma, cerebral amyloid angiopathy, intracellular neurofibrillary tangles (NFT) composed of the microtubule-associated protein tau, neuropil threads, and dystrophic neurites containing hyperphosphorylated tau (Fig 1.2, 1.3) (Crews and Masliah 2010). In addition to these pathological processes, AD sufferers display a characteristic loss of neurons, loss of synaptic function and dendritic arborisation. Moreover an increase in neuroinflammation and brain atrophy in cortical and subcortical regions has been demonstrated (Mayeux and Stern, 2012; Xu et al., 2013). The cerebral areas most severely impacted are the entorhinal cortex, 
 hippocampus and the basal forebrain.
hippocampus and the basal forebrain.
AD is hallmarked by a progressive deterioration in multiple cognitive domains, severe enough to result in a global alteration or loss of cognitive ability. This manifests ante-mortem as the following behavioural symptoms; memory impairment, aphasia, temporal and spatial disorientation, poor judgment, delusions, sleep deprivation, weight loss, and changes in mood and personality (Guillot-Sestier and Town; 2013). Behavioural symptoms correlate with regions of pathology and brain atrophy. As is common with all progressive neurodegenerative diseases, behavioural symptoms will worsen as pathology increases.
1.5.1 Production of Amyloid Beta
Amyloid beta is a 36-43 amino acid polypeptide derived from the cleavage of a membrane glycoprotein known as Amyloid Precursor Protein (APP) (kang et al., 1987). The APP gene is located on chromosome 21 and consists of 18 exons which span 290 kilobase pairs (kbp) (Bergsdorf et al., 2000). Aβ is encoded by exons 16 and 17 of the APP gene. In humans, alternative splicing of APP mRNA leads to the generation of isoforms which express proteins of between 639-770 aa. The major APP isoforms expressed in the brain are APP695, APP751 and APP770. Neurons preferential express the APP695 isoform, whilst APP751 is the primary APP isoform outside the CNS (). Though each isoform is capable of generating Aβ, its secretion is by far highest by Neurons and therefore the APP695 isoform. (Cirrito et al., 2005)
APP is a highly conserved multifunctional protein consisting of a large extracellular N-terminal domain, a transmembrane domain and the APP intracellular domain (AICD) at its C terminus (Rohan De Silva et al., 1997). Aβ is derived from the sequential proteolysis of APP by the aspartyl proteases α or β, followed by γ–secretase. The first form of APP cleavage produces the protein p3 from sequential cleavages by α- and γ-secretases in what is called the non-amyloidogenic pathway. Whereas in the amyloidogenic pathway, cleavage of APP by β- and subsequently γ-secretase generates the Aβ released into the neuronal milieu (Schaefer et al., 2011).
These secretases cleave Aβ within the luminal domain, The main neuronal β- secretase is a transmembrane aspartyl protease known as BACE1. This cleaves APP within its ectodomain, leading to the N-terminus of Aβ. This creates a 99 amino acid CTF and cleavage by the γ-secretase leads to generation of a 50 amino acid cytoplasmic polypeptide known as APP intracellular domain (AICD) (Sisodia 2007; Edbauer et al. 2003). The isoform of Aβ produced by this cleavage is dependent on the location of the cleavage site.The non-amyloidogenic pathway accounts for 90% of APP processing and APP is cleaved within the Aβ region (at Lys16-Leu17) and not only does α-secretase stop the formation of the amyloidogenic protein but the large ectodomain released from APP has been shown to have a neuroprotective and memory enhancing function (Furukawa et al., 1996; Nalivaeva, Turner 2013; Allinson et al. 2003). α-secretases include tumour necrosis factor-α-converting enzyme (TACE) and a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) as well as other metalloproteases (Caescu, Jeschke & Turk 2009). Once the N terminal of the protein has been generated through α or β secretase activity, the remaining part of APP can now be cleaved. This is brought about by an intramembranous cleavage of α or β CTFs by γ-secretase activity, which liberates the p3 (3kDa) protein from the α CTF and Aβ (4kDa) is released into the extracellular space from the β CTF (Sisodia 2007; Edbauer et al. 2003).
Image(here).
1.5.2 Normal function of Amyloid Beta
The function of Aβ has been investigated by numerous researchers from a pathological point of view. However, it is important to note that Aβ also carries out normal physiological functions, though their role requires further elucidation. Cell culture experiments conducted by Plant and colleagues treated rat and human cortical neurons with inhibitors of γ- or β-secretase as well as an antibody of Aβ. Neuronal viability as measured via an MTT assay was significantly reduced in these experiments, however when cells were co-incubated with Aβ cell viability was restored (Plant et al. 2003). Thus, indicating that one of the primary functions of Aβ is the maintenance of cell viability. However, the exact mechanism by which it maintains cell viability has yet to be clarified. One suggestion is that Aβ functions as a negative feedback mechanism to modulate neuronal activity (Ting et al. 2007; Kamenetz et al. 2003).
It has also been hypothesised that Aβ plays a role in synaptic plasticity and thus mediates learning and memory formation (Huber et al. 1993). Rodents injected with competitive inhibitors of endogenous Aβ prior to a learning exercise showed a severe disruption in their ability to acquire and retain memory up to 5 days post training. Indicating that one major pre-disease function of Aβ is the formation of memories. Further corroborating with this hypothesis, intrahippocampal treatment with Aβ after learning exercises significantly enhanced retention of memories (Garcia-Osta et al., 2009).
1.6 Genetics
Genetics is another risk factor that shows an increased propensity towards the development of AD. About 5% of AD sufferers are genetically predisposed towards the disorder due to Mendelian autosomal dominant patterns of inheritance and are thus said to suffer from Familial Alzheimer’s disease (Bertram et al., 2010; Philipson et al., 2011; Schellenberg and Montine, 2012). The onset of disease phenotype for these patients is between 40-50 years of age (Zhang et al., 2012)
1.6.1 Early onset Alzheimer’s disease
Due to genetic studies of individuals with early-onset familial dementia, it was discovered that a majority of these individuals had mutations in at least one of three genes directly involved in amyloid production. First was the transmembrane amyloid precursor protein (APP) on chromosome 21. As oligomers of Aβ42 were discovered to be more prone to aggregating than Aβ40 and were also shown to correlate best with neurotoxicity and cognitive dysfunction, many researchers were led to posit that mutations in the genes coding for both APP, or α, β, and γ–secretases may play a significant role in the development of AD (Xu et al., 2013). Genetic studies, geared towards unearthing the cause of AD identified mutations in the genes encoding presenilin 1 (PS1) and 2 (PS2), PSEN 1 and PSEN 2 respectively. PS1 and PS2 are proteins, which form the catalytic core of the γ-secretase complex (Goate et al., 1991, Levy- Lahad et al., 1995, Sherrington et al., 1995, Giasson et al., 2003).
1.6.2 Late onset Alzheimer’s disease
Remarkably, late-onset AD (LOAD), often defined as sporadic in nature, has been shown to have a strong genetic component. Genome-wide association studies (GWAS) have identified novel loci involved in lipid metabolism, immune response, endocytosis, tau metabolism, axonal development and epigenetics, which contribute towards AD risk. Many of these genetic polymorphisms contribute mild to high AD risk and even more importantly determine the time of onset. The major genetic point of interest for LOAD is Apolipoprotein E (APOE), a regulator of cholesterol transport, involved in neuroplasticity, tau phosphorylation and inflammation. The presence of the ε4 allele of the gene for Apolipoprotein E (APOE) is the largest genetic risk factor for the development of sporadic AD. Between 40 and 80% of people with AD possess at least one APOEε4 allele. “One copy of APOE ε4 increases the risk for AD threefold and two copies further elevate the risk twelvefold” (Corder et al., 1993; Huang and Mucke, 2012).
In AD, APOE binds to Aβ and functions as a catalyst that enhances the proteolysis of Amyloid Beta, and thus prevents its abnormal aggregation (Kim et al., 2009; Castellano et al., 2011). Moreover, binding of APOE to the low-density lipoprotein receptor-related protein 1 (LRP1) on the plasma membrane of astrocytes regulates the metabolism of soluble Aβ (Verghese et al., 2013). Neuropathological, Neuroimaging and transgenic mice studies have all shown a higher deposition of Aβ in APOε4 positive carriers than in the ε4 negative counterparts. Despite these facts, APOE is not considered a causative gene, as many develop AD with or without its contribution. Rather it has been suggested that it is a determinant of age of onset.
Several genes associated with neuro-inflammation have been identified in GWAS. Single nucleotide polymorphisms in the gene CR1, which encodes the complement (3b/4b) receptor 1 were identified in individuals with LOAD. The CR1 protein is widely expressed on the surface of erythrocytes, microglia, and neurons resulting in the phagocytosis of complement opsonised particles. It modulates the effect of APOE ε4 on brain fibrillar amyloid burden and can bind to C3b and C4b, thus moderating the activity of the complement system (Rogers et al., 2006). Pathological studies have demonstrated that there is a correlation between the elevated expression of CR1 mRNA and AD, which is seemingly at odds with the fact that CR1 facilitates Aβ clearance. However, the identified polymorphisms encode high-expression (1400 copies per cell) and low-expression (<200 copies per cell) alleles (Krych-Goldberg et al., 2002). High levels of CR1 protein may exacerbate the complement cascade whereas; lower levels may lead to impaired clearance of Aβ. Thus, the increase in mRNA expressions of either polymorphism may have different effects, which contribute in their own way towards AD pathology and increase the risk of developing the disease.
Other genes of note include Clusterin, an apolipoprotein upregulated in AD which influences fibril formation, Aβ clearance, neurotoxicity and regulates the complement membrane attack complex; ATP-binding cassette transporter A7 (ABCA7), upregulated 10 fold in microglia, modulates microglial phagocytosis of apoptotic cells and Aβ via the complement pathway; CD33 expressed in microglia, which is positively correlated with insoluble Aβ42, plaque burden, cognitive decline and has been postulated to inhibit Aβ phagocytosis; and the Triggering receptor expressed on myeloid cells 2 (TREM2), thought to impair the ability of brain leukocytes to maintain a homeostatic balance of amyloid beta (reviewed in Karch and Goate, 2014).
1.7 Causal Hypothesis
1.7.1 Amyloid cascade hypothesis

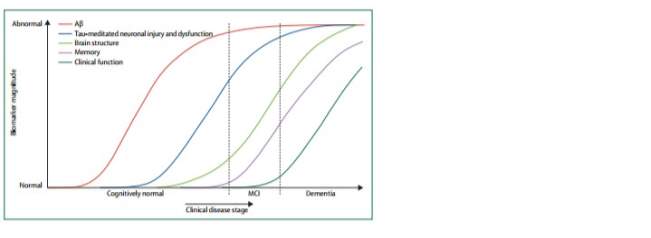 The most popular theory is the amyloid cascade hypothesis (Hardy and Allsop 1991; Karran et al., 2011). The initial hypothesis posits that amyloid deposition, results in the initiation of AD with numerous secondary processes – such as the hyper-phosphorylation of the microtubule-associated protein tau (MAPT), and the formation of NFT occurring subsequently. Accumulation of these deposits then leads to neuronal dysfunction and eventually dementia (Selkoe, 1996). Several observations made in the late 1980s and early 90s corroborate with this hypothesis. First, was the detection of Aβ as an important component of the senile plaques (Glenner et al., 1984), which Alzheimer described in 1907. Genetic studies, geared towards the proof of this hypothesis, identified familial mutations in APP on chromosome 21, PS1 and PS2; thus providing an added boost towards the amyloid cascade hypothesis (Goate et al., 1991, Levy-Lahad et al., 1995, Sherrington et al., 1995, Giasson et al., 2003). Furthermore, individuals with Down syndrome – a condition hallmarked by the triplicate of chromosome 21- display many of the same pathological hallmarks and cognitive deficits as those who suffer from AD. Following the amyloid cascade hypothesis, and recent amyloid imaging studies, it is thought that Aβ accumulation begins first (Jack et al., 2011). Aβ exists in a number of different conformations in the CNS such as, monomers, oligomers, protofibrils, fibrils and Aβ plaques. The degree to which they promote pathology differs. Deposition of Aβ is thought to be succeeded a tau-mediated neuronal injury, brain atrophy, and cognitive decline (Mintun et al., 2006, Bourgeat et al., 2010). Jack and colleagues (1999, 2007, 2011) proposed a hypothetical timeline for the progression of the disorder based on the use of biomarkers (Fig1.4).
The most popular theory is the amyloid cascade hypothesis (Hardy and Allsop 1991; Karran et al., 2011). The initial hypothesis posits that amyloid deposition, results in the initiation of AD with numerous secondary processes – such as the hyper-phosphorylation of the microtubule-associated protein tau (MAPT), and the formation of NFT occurring subsequently. Accumulation of these deposits then leads to neuronal dysfunction and eventually dementia (Selkoe, 1996). Several observations made in the late 1980s and early 90s corroborate with this hypothesis. First, was the detection of Aβ as an important component of the senile plaques (Glenner et al., 1984), which Alzheimer described in 1907. Genetic studies, geared towards the proof of this hypothesis, identified familial mutations in APP on chromosome 21, PS1 and PS2; thus providing an added boost towards the amyloid cascade hypothesis (Goate et al., 1991, Levy-Lahad et al., 1995, Sherrington et al., 1995, Giasson et al., 2003). Furthermore, individuals with Down syndrome – a condition hallmarked by the triplicate of chromosome 21- display many of the same pathological hallmarks and cognitive deficits as those who suffer from AD. Following the amyloid cascade hypothesis, and recent amyloid imaging studies, it is thought that Aβ accumulation begins first (Jack et al., 2011). Aβ exists in a number of different conformations in the CNS such as, monomers, oligomers, protofibrils, fibrils and Aβ plaques. The degree to which they promote pathology differs. Deposition of Aβ is thought to be succeeded a tau-mediated neuronal injury, brain atrophy, and cognitive decline (Mintun et al., 2006, Bourgeat et al., 2010). Jack and colleagues (1999, 2007, 2011) proposed a hypothetical timeline for the progression of the disorder based on the use of biomarkers (Fig1.4).
Neuroinflammation is a chronic inflammation of the central nervous system triggered by trauma and infections to the CNS. Markers of CNS inflammation include increased activation of microglia, pro-inflammatory cytokine concentration, blood-brain-barrier permeability, and leukocyte invasion. Over the past 20 years, inflammation in the CNS has been extensively studied both as a cause of neurodegeneration, though more importantly as a potential target for novel therapeutics.
The hypothesis that post Aβ pathology in AD is caused by neuroinflammation has recently gained traction. The amyloid cascade-inflammatory hypothesis posits that while the abnormal production and aggregation of Aβ acts as a trigger, it is not in itself sufficient to be causative of the disease (McGeer and McGeer, 2013). Rather it is thought that the presence of aberrant deposits of Aβ may induce inflammation. In turn, inflammation may then enhance tau up-regulation and promote tau aggregation. These postulations are largely due to two major observances: non-steroidal anti-inflammatory drugs reduce the risk of developing AD and; several lifestyle/environmental factors which are known risk factors for the development of AD display an inflammatory component. For example, the hypothesis suggests that ageing – the single largest risk factor for the development of AD – is influential due enhanced chronic inflammation associated with increasing age (Blasko et al., 2004). Moreover, many of the aforementioned risk factors for AD such as obesity and systemic disease have been shown to display an increase in the concentrations of inflammatory mediators. Studies have suggested that systemic and chronic conditions are associated with increased cognitive decline, hippocampal atrophy (Yaffe et al., 2004; Marsland et al., 2008),changes in electroencephalograph readings (Semmier et al., 2012) and an increased risk of Alzheimer’s disease (Engelhart et al., 2004; Tan et al., 2008, Bermejo et al., 2008). This cognitive decline was positively correlated with the expression of the pro-inflammatory mediator TNFα (Holmes et al., 2009).
Numerous recent studies have indicated that the presence of extracellular Aβ deposits is considered to be an important trigger for the chronic neuroinflammation that has been observed in AD brains. This is based on the observation of activated microglia, increased levels of complement elements, cytokines and chemokines in regions surrounding amyloid plaques. The pro-inflammatory enzyme COX-2 is elevated early in AD progression, and IL-6 and transforming growth factor-β1 (TGF-β1) were also found to be elevated in patients with severe dementia (Luterman et al., 2000; Ho et al., 2001; Pasinetti, 2001). Moreover, a long-term epidemiological study demonstrated that the use of non-steroidal anti-inflammatory drugs (NSAIDs) lowers the risk of AD (Etminan et al., 2003). Ibuprofen and indomethacin have been shown to decrease the levels of secreted Aβ42 in vitro as well as in animal models of AD (Weggen et al., 2001; Eriksen et al., 2003). As the levels of Aβ40 have remained unaffected, it is likely that NSAIDs are causing modulation of either APP or gamma secretase (Kukar et al., 2008).
Not all conventional NSAIDs provide benefits (Aisen et al., 2003; Kukar et al., 2005). Some NSAIDS have been shown to accelerate AD pathogenesis in patients with advanced progression (Breitner et al., 2009). It is therefore apparent that the effects of NSAIDs may provide beneficial or detrimental effects dependent on the progression stage of the disease and the type of NSAID used. This dependence may explain failure of NSAIDS as a treatment for AD in many clinical trials. Therefore, the pathways by which beneficial NSAIDs reduce specific inflammatory mechanisms, and thus how those inflammatory processes contribute to the pathogenesis of AD must be identified.
1.8 Current therapeutic strategies for the treatment of AD
1.9 Cellular components of the Central Nervous System
1.9.1 The blood brain barrier
An optimally functioning CNS is a well-calibrated chemical, ionic and anti-inflammatory environment ideally suited to ensure optimal neuronal function. The blood-brain barrier (BBB) is a physical barrier of phylogenetic origin which restricts molecular transport between peripheral and CNS vasculature, thus protecting the neuronal milieu from peripheral innate and adaptive immune cells, plasma proteins and oscillations in the concentration of inflammatory mediators which would offset the delicate homeostatic balance of the brain(Sharma et al., 2012). Structurally, the BBB defines the tight junctions that connect endothelial cells of the brain capillaries. Endothelial-lined capillaries form a core, which is in turn surrounded by astrocytic end- feet, a basal lamina, and pericytes (Marques et al., 2013). Every neuron is perfused by its one such capillary and a dynamic crosstalk exists between the BBB endothelial cells, glia and neurons thus highlighting how essential a functioning BBB is in the maintenance of neurons and prevention of aberrant disease like states (Abbott et al., 2006). Moreover, degeneration and increased permeability of the BBB has been well characterised in many of the AD-associated risk factors further highlighting its importance.
BBB permeability increases with the progression of age. Similarly, in AD, BBB permeability is more pronounced and grows at an accelerated rate when compared to the progression of permeability in age-matched controls. Post-mortem studies have shown BBB damage in AD including BBB-endothelial cell necrosis, increased exocytosis, cortical and in particular hippocampal accumulation of blood-derived proteins such as immunoglobulins, albumin, fibrinogen, serum amyloid P and thrombin (Fiala et al., 2002; Zipser et al., 2007; Hultman et al., 2013) and degeneration of BBB- pericytes (Sengillo et al., 2013).
The endothelial cells, which form an integral part of the BBB, express receptors for pro-inflammatory mediators on their cell membrane. Exposure to peripheral inflammatory ligands such as TNF, IL-1β and IL-6 may elicit barrier-opening, lead to an influx of inflammatory mediators and may alter the normal response of the brain to various stimuli. High local concentrations of the anti-inflammatory mediators TGF-β, IL- 10, and gangliosides, which are toxic to T cells, make the CNS a robust environment capable of withstanding inflammation to a degree (Irani et al, 1996; Strle et al., 2001; Malipiero et al., 2006). However, with age and other peripheral systemic states there is an increased BBB permeability. Thus, the BBB becomes more susceptible to inflammation and if damaged enough may lead to an influx of peripheral immune cells into the CNS. Therefore, despite the fact that the CNS is not an immune privileged organ, it may be that systemic triggers lead to receptor activation of a failing BBB and are required to initiate neuroinflammation. Chronic perpetuation of which may result in chronic neuroinflammation and initiate many of the pathogenic neurodegenerative states of the CNS.
A few studies suggest that BBB failure may initiate neuroinflammation and AD due to an inadequate efflux-influx balance. For example, it is thought that the influx of serum amyloid protein and plasma amyloid through the BBB may be the point of origin for Aβ aggregation (Pluta, 2007). The receptor for advanced glycation end products (RAGE) – present on BBB-endothelial cells – is of particular note, as binding to Aβ or its natural ligand AGE can activate it. Ligand binding to RAGE leads to the activation of NF-κB, downstream of which causes changes to the expression of inflammatory mediators and an increase in RAGE expression. In transgenic models of AD and β-amyloidosis, RAGE expression is elevated in neurons, microglia and vasculature proximal to sites of pathology (Yan et al., 2006; Deane et al., 2003). Particularly noteworthy since many individuals living with AD risk factors also display elevated levels of AGE (Vlassara et al., 2002; Goldin et al., 2006). Thus, RAGE activation and the resulting influx of peripheral proteins leads to a chronic cycle of activation and is a likely contributor towards the chronic neuroinflammatory state observed in AD.
Conversely, impaired efflux of Aβ has also been observed in AD. Expression of LRP-1 the main transporter that mediates efflux of Aβ is reduced in AD patients (Donahue et al., 2006). Hence, despite the opposing roles of RAGE and LRP-1 it is evident that both contribute towards the homeostatic balance of the CNS. Changes in the expression of both leads to the presence of amyloid protein in the CNS, which sustained may result in the pathogenesis of AD. However, it is unclear from these studies whether the BBB impairment is causative in nature, or whether it exacerbates a pre-existing neurodegenerative/neuroinflammatory condition.
.
1.9.2 Neurons
Neuronal apoptosis leading to regional atrophy of the cortical regions is characteristic of neurodegenerative diseases. Originally thought to be unfortunate bystanders and unwitting “victims” in the degenerative process, neurons are capable of producing inflammatory mediators that are damaging to the neuronal milieu. They are one source of complement proteins, COX, IL-1β, IL-6 and TNF-α a majority of which are pro-inflammatory in nature (reviewed in Heneka et al., 2007). Also, neurons have been shown to express iNOS (inducible nitric oxide synthase). In the long-term release of NO – a chemical known to cause neuronal
dysfunction and apoptosis – will be damaging (Heneka et al., 2001). It is possible that TNF-α and NO in low concentrations may have a neuroprotective effect, but this theory is not well characterised (Heneka et al., 2007).
1.9.3 Astrocytes
Astrocytes are glial cells with fine elongated processes, giving the cells a star-like appearance. Though these are the most abundant cells in the CNS, each cell occupies a distinct territory. A territory contains up to 2,000,000 neuronal synapses – with the only astrocyte- astrocyte interaction occurring between the end-feet of the astrocytic processes (Bushong et al., 2002; Oberheim et al., 2009). This interdigitation forms gap junctions, connecting adjacent astrocytic cells, thus building an entire network with close contact to neurons, synapses and the cerebral vasculature (Sofroniew et al., 2010). Functionally, astrocytes are involved in synaptic formation, maturation and pruning during the developmental stages. Post- development, astrocytes maintain the ionic milieu of the CNS, regulate long-term potentiation, facilitate neurotransmitter release and uptake, modulate the passing of information at the synapse, establish bi-directional communication with neurons, and control the BBB via their perivascular end-feet (Clarke and Barres, 2013).
Astrogliosis is a process, which results in the conformational change (Hypertrophic soma and processes) of reactive astrocytes, an increase in expression of glial fibrillary acidic protein (GFAP) intermediate filaments and, increased proliferation. Aβ may activate astrocytes, and thus elicit a neuroprotective or neuro-detrimental change in normal function of astrocytes. Post-mortem tissue samples of individuals with AD show activated astrocytes clustered at sites of Aβ deposition and astrogliosis (Wisniewski and Wegiel, 1991). The large group of astrocytes proximal to Aβ deposition sites indicates that once activated astrocytes generate chemotactic molecules, which result in the further recruitment of other astrocytes to the site. A subsequent study found a positive correlation between phagocytosed Aβ within astrocytes of the entorhinal cortex and the amount of AD pathology in the local milieu. ApoE polymorphisms may increase the risk of developing AD by affecting phagocytosis of Aβ (Nino et al., 2001). While there is a definite role for astrocytes in the clearance of Aβ it appears that they may also contribute to the generation of Aβ and thus a neuro-detrimental pathway.
Β-Secretase (BACE1) known to result in the cleavage of the Aβ42 isoform, is over- expressed when exposed to chronic stressors. Therefore, it is possible that activation of astrocytes by inflammatory mediators and or Aβ may result in the generation of Aβ thereby perpetuating the condition. Granting credence to this theory, an APPV717I study demonstrated that transgenic mice display glial activation prior to the local deposition of Aβ (Nunomura et al., 2001). Further studies posit a neuro-detrimental role for astrocytes in AD. Oscillations in calcium signaling were observed in those with the Familial AD presenilin 1 (PSEN1) mutation (Haughey and Mattson, 2003). It is likely that aberrant calcium signalling may contribute to much of the neuronal dysfunction, neuronal apoptosis and disintegration of the synapse observed in AD. Finally, when present, astrocytes inhibit the ability of microglia to phagocytose Aβ deposits either by creating a physical barrier or by releasing proteoglycans (Akiyama et al., 2000). Astrocytes express some inflammatory mediators including complement subcomponents and receptors, IL-1β, IL-6, COX, TNF-α and prostaglandins (reviewed in Akiyama et al., 2000).
1.9.4 Microglia
Microglial cells known as the resident macrophages of the CNS are key mediators of the immune response. These cells are derived from progenitors of myeloid origin, which migrate from the periphery and enter the brain during early development. Postnatally, they are found to be disseminated through the brain, primarily in a “resting” ramified phenotype, characterised by a small soma with fine cellular processes. Microglial processes are equipped with neurotransmitter, cytokine, chemokine and pattern recognition receptors which function to sense pathogen-associated molecular patterns (PAMPs) and host-derived danger associated molecular patterns (DAMPs), such as misfolded proteins, mislocalised nucleic acids and protein aggregates as found in AD, and to respond to these signals by activating and adopting a change in morphological and functional phenotype. Brain-derived neurotrophic factor, essential for the formation of learning-dependent synapses and thus the development of memory by long-term potentiation is secreted by microglia.
Microglia play a role that aims for neuroprotection by the elimination of Aβ aggregates via phagocytosis (Heneka et al., 2010). However, microglia tend to have more detrimental than beneficial effects by contributing to neuronal cell death and neurodegeneration. Pathological events such as infection, ischaemia, traumatic injury, tumour invasion, neurodegenerative diseases and changes in the homeostatic balance of the CNS may present some of these DAMPSs and can result in microglial activation. Activated/Reactive microglia can be characterised by a reduction in cell processes, amoeboid appearance, up-regulation of gene
expression of surface receptors and function. Persistence of pro-inflammatory mediators – as found in states of chronic neuroinflammation – can cause microglia to remain activated for extended periods, releasing quantities of cytokines and neurotoxic molecules that contribute to long-term neurodegeneration (Liu and Hong, 2003). This functional perturbation of microglia from its normal neuroprotective states may exacerbate the progression of pathogenesis in many neurodegenerative states.
There is a strong signalling overlap between pathways activated by external pathogen associated molecular patterns (PAMPS) and the host-derived DAMPs, such that microglia may recognize and react to host aggregated proteins with the same exaggerated response as to an invading pathogen. Amyloid β can bind to several innate immune receptors expressed on microglia, such as NOD-like receptor (NLR), CD14, CD36, CD47, α6β1 integrin, class A scavenger receptor, RAGE and toll-like receptors (TLRs) 2, 4, and 6, all of which can lead to activation when triggered. Initial activation of microglia by Aβ results in the chemotaxis and recruitment of more microglial cells to sites of Aβ deposition (Terry and Wisniewski, 1975). In one study AD mice with Cx3cr1 (a chemokine receptor) had high levels of microglial migration; whereas Cx3cr1-/- mice had neither microglial accumulation nor neuronal loss, in addition with low levels of hyperphosphorylated tau (Osherovich, 2008). Knock-out animal models for the scavenger receptors A and B present on microglia which promote clearance of Aβ have shown increases in the amyloid burden. Evidently microglia play one of the most crucial roles in the perpetuation of the neurodegenerative and chronic inflammatory state that exists in AD.
Activation of microglia by Aβ also results in the expression of pro-inflammatory cytokines such as IL-1β, and IL-18 in their inactive forms. Cleavage into pro/active forms is controlled by a large molecular signalling complex known as the inflammasome, which has particular significance in the development of chronic inflammation. As such its components, which include the cleavage molecule caspase 1, adaptor protein ASC, the NLRP3 domain (sensor NLR, LRR- and pyrin domain-containing 3); are of particular interest as regions of possible therapeutic benefit.
1.10 The immune system in AD
The human immune system is an intricate system of immune sensors and mediators, which acts as the first line of defence against infection, injury and invasion by pathogens such as viruses, bacteria and fungi. It is composed of the innate and acquired (adaptive) immune responses. Of the two, innate immunity is the phylogenetic predecessor, whereas adaptive immunity is an evolutionary phenomenon that can be attributed only to vertebrates.
Innate immune responses are rapid (0-12 hour) responses to antigens that are reliant on pattern recognition receptors (PRRs) present on innate immune cells (macrophages, neutrophils and dendritic cells); and inflammatory mediators, both passed through the organism’s germ line (Murphy, 2012). Immunity of this sort acts in a non-specific manner towards antigens and can neither be acquired nor adapted during an organism’s lifetime. Conversely, adaptive immunity is an acquired form of immunity that is developed in response to primary exposure to antigens. Its response is extraordinarily discriminative, and its components “adapt” to the presence of pathogens by activating, proliferating, and creating potent mechanisms for the removal of pathogens. It further differs from the innate immune response by the slow duration of response and its immunological memory.
On presentation of an antigen, components of the innate immune system such as lysozymes, defensins, mucin, lectin, macrophages and the complement system first act to lyse and phagocytose pathogens. In the second phase, PRRs expressed on innate immune cells recognize non-self motifs on host cells- known as pathogen-associated molecular patterns and activate. Activation sets off a cascade of effector mechanisms, ultimately resulting in the removal of the pathogen. However, the innate immune system can be overwhelmed or evaded by pathogens. In such a scenario where the first two lines of defence are breached, innate immunity can trigger the adaptive immune system.
The complement system is a crucial component of the innate immune system. Complement first discovered by Jules Bordet is defined as a “heat-labile component of normal plasma. It is comprised of approximately 40 plasma/serum and cell-membrane-associated glycoproteins. Although complement proteins are found predominantly in plasma, a disruption of the blood brain barrier is not required for complement to be present in the CNS. In response to CNS changes microglia, glial cells and neurons can synthesise complement proteins (Gasque et al., 2000). Their role is to facilitate the removal of pathogenic organisms via four primary functions: recognition, opsonisation, inflammatory stimulation, and direct killing through the membrane attack complex (Maibaum, 2000). The complement membrane attack complex(MAC) forms transmembrane channels that penetrate through the phospholipid bilayer of the target cells, leading to cell lysis and apoptosis.
Initiation of the complement cascade can be achieved by either the Innate or the Adaptive immune system via one of three pathways: the C1q pathway (classical pathway), an amplification loop (alternative complement pathway) and the lectin pathway (Fig 1.5). Upon activation of the complement system by several triggers, many inflammatory mediators are produced, initiating inflammatory processes. If unregulated, this cascade will result in an amplification of the inflammatory response and cell apoptosis.
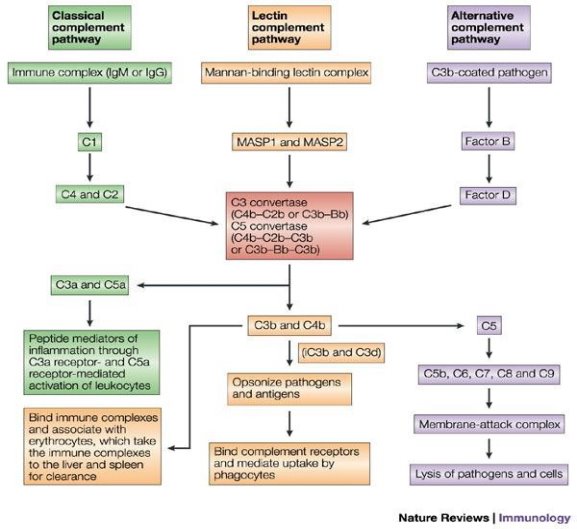

The classical pathway involves the activation of the C1 complex, composed of the glycoproteins C1q, C1r and C1s associated in a calcium-dependent macromolecular complex. C1r and C1s are serine proteases activated primarily by the interaction of C1q protein with antibody-antigen complexes containing IgG or IgM (Kishore and Reid, 1999). Activation can also occur subsequent to the interaction of C1 with molecules such as serum amyloid P, DNA, RNA and C-reactive protein (Sim and Malhotra, 1994; Gaboriaud et al., 2003). Once a ligand has bound to C1q, a conformational change occurs in the collagen rich region. This change leads to the activation of C1r and eventually C1s. C1s acts to cleave the complement proteins C2 and C4 into C2a, C2b, C4a and C4b respectively, resulting in the formation of a C4bC2a complex known as C3 convertase.
Activation via the lectin pathway occurs when pathogens are presenting the carbohydrate antigen mannan on their surface bind to mannose-binding lectin (MBL) and ficolins. Once bound, proteases associated with MBL known as MBL-associated-serine-protease (MASP) can also cleave C2 and C4 complement proteins, resulting in the formation of C3 convertase.
Alternative pathway activation occurs via the spontaneous hydrolysis of C3. This slight conformational change in the protein enables the binding of factor B and results in its subsequent cleavage into subunits Ba and Bb. Similarly, to the classical pathway of complement activation, the result is the production of a C3 convertase complex, this time composed of C3bBb.
All three pathways converge at the formation of C3 convertase. The function of C3 convertase is to cleave and activate component C3, creating C3a and C3b. C3a is a weak chemotaxis agent, with the primary role of microglial and phagocyte recruitment (Kishore et al., 2003). While the C3b fragment attaches to the pathogen’s surface and can act as an opsonin for target recognition and subsequent phagocytosis. Also, C3b binds with the C3 convertase to form C5 convertase (C4b2a3b). Subsequently C5-convertase cleaves C5 into the anaphylatoxin C5a and C5b the base for the formation of the MAC (C5-C9 complex). The MAC creates pores in the lipid bilayer of pathogens or host-derived cells in chronic inflammatory states causing cell lysis and apoptosis (Shastri et al., 2013). Amplification and or chronic activation of the complement cascade will inevitably lead to tissue atrophy.
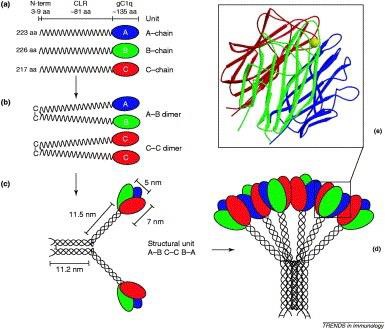
C1q, the recognition subcomponent of the classical complement pathway, is a 460kDa protein consisting of 18 polypeptide chains in 3 subunits of 6 (6A, 6B and 6C) (Fig 1.6). Chiefly produced by cells of myeloid origin, microglia are the only CNS resident cells capable of synthesising C1q (Lynch et al., 2004). Each chain consists of a 3-9 base pair (bp) N-terminal region, an 81 bp collagen-like region (CLR) and the approx 135 bp C-terminal globular head region (gC1q) (Sellar et al., 1991). Interchain disulphide bonds between N-terminal cysteine residues yields dimer subunits of 6 A-B and 3 C-C. The CLRs of the A-B and C-C subunits then bind through covalent and non-covalent bonds to give rise to a triple helical collagen-like unit ABC-CBA. Three of these units bind further via non-covalent bonds to form the hexameric C1q, which has been described as having morphology similar to a” bouquet of tulips” (Reid and Porter, 1976; Kishore and Reid, 1999; Kishore et al., 2003).
The globular gC1q domain (ligand recognition domain) is a heterotrimeric structure, formed from the C-terminal regions of the A, B and C chains (Kishore et al., 2003; Nayak et al., 2010). These modules are denoted globular heads A (ghA), B (ghB) and C (ghC). Proximal to the apex of the gC1q domain lies a calcium ion, which plays a significant role in the binding of C1q to immune complexes (Roumenia et al., 2005).
Protein crystallisation studies have also revealed a three-dimensional structural similarity between the gC1q domain and tumour necrosis factor (TNF), hence leading to recognition of a C1q/TNF superfamily. The members of this family are crucial in a range of functions including inflammation, adaptive immunity, energy homeostasis and tissue regeneration (Kishore et al., 2004). The amino acid similarity (ranging from 20-96%) between family members has been found conserving eight invariable residues, critical in the maintenance of the structural integrity of the gC1q domain. Thus, this highlights the importance of jelly roll topology throughout evolution in regard to C1q’s various functions (Nayak et al., 2011). On the other hand, C1q overall structural organisation is similar to the MBL, surfactant protein A (SP-A), surfactant protein D (SP-D) and ficolin, which are PPRs belonging to the collectins family. (Nayak et al., 2011). The ability to express these modules as recombinant vectors has been enlightening in the understanding of how C1q is activated by a host of self and non-self-ligands. A study by Kishore and colleagues (1998) revealed that although IgG and IgM seemingly bind with the same affinity to C1q, ghB preferentially binds to IgG. This preferential binding was due to the fact that ghA and ghC show a combination of basic and acidic residues on their external surfaces whereas ghB shows a predominantly positive charge on its external face due the presence of Arginine residues. Further studies have elucidated that ghA can bind heat- aggregated IgG, IgM and HIV-1 gp41; ghB binds to heat-aggregated IgG as well as amyloid β and; ghC shows a preference for IgM and HTLV-1 gp21 peptide (Kishore et al., 2004). Thus, globular heads are not only structurally different but also functionally autonomous. Consequently, the multivalency of C1q’s function is due to its heterotrimeric structure.

The primary function of C1q is the recognition of immune complexes, thus initiating the classical complement pathway (Kishore et al., 2003). Moreover, it is involved in a varied number of processes such as phagocytosis of bacteria, neutralisation of retroviruses, cell adhesion, modulation of dendritic cells (DC), B cells and fibroblasts, and the clearance of apoptotic cells (Kishore et al., 2004). Due to its ability to be activated by a broad range of self and non-self-ligands – such as IgG, IgM, A, serum amyloid P, HLTV1, HIV gp41, Cd91], Lipopolysaccharide, gram-negative bacteria, phospholipids, pentraxins and C-reactive protein- in an extensive range of diseases, it’s overall significance is second only to that of complement subcomponent 3b (Nayak et al., 2010).
Copious evidence indicates that C1q and the complement system play a role in the pathophysiology of Alzheimer’s disease. AD and the complement system became linked by the discovery by Rogers et al. 1992 that Aβ binds to complement protein C1q in-vitro. In regions of the brain with a high amyloid load, the expression of C1q is up-regulated 80-fold compared to control levels (Yasojima et al., 1999). C1q synthesis is induced in neurons during ageing, and subsequent to Aβ treatment. In particular the mRNA expression of the globular head B of C1q has been shown to be significantly higher in AD than in control. A fact confirmed by the specific affinity of ghB for Aβ (Kishore et al., 2003). Further correlating with these studies, immunohistochemical staining of human and rodent post-mortem samples has shown C1q to be co-localised with fibrillar AB (thioflavin-S) plaques and detected the presence of the C5b-9 MAC when compared to controls (Tenner and Fonseca, 2006). Although these studies introduce a role for C1q in AD, later studies have only just begun to characterise what this role is and the part it plays in the pathogenesis of AD.
It has been postulated that C1q plays a neurodetrimental in the CNS. One study identified by Webster and colleagues identified a contribution of C1q towards the aggregation of AB. In-vitro experimentation demonstrated a 7-fold enhancement of Amyloid beta aggregation on co-incubation with C1q (Webster et al., 1994), indicating that C1q may be a trigger for the aggregation of AB into fibrillar amyloid. Previous in-vivo experimentation has shown fibrillar amyloid aggregation, to be one of two major focal points at which clinically significant cognitive decline occurs (Reviewed in Tenner and Fonseca, 2006). Therefore, it can be postulated from this evidence that C1q is a major contributor towards both the pathology (fibrillar amyloid plaques) and the resulting behavioural (cognitive decline) observed in AD.
Corroborating with the studies mentioned above, C1q-/- transgenic mice over- expressing an aberrant form of APP, were created to characterise the role of C1q in AD. When compared to transgenic mutant APP mice with the comprehensive complement system, there was a significant decrease in the observed inflammatory glial markers proximal to the plaques in C1q-/- mice. In addition, the researchers further reported an improvement in the neuronal integrity of APP C1q-/- when compared with APP mice, despite there being no change in the plaque pathology (Fonseca et al., 2004). Furthermore, inhibiting the binding of C1q to Aβ may lead to the protection of hippocampal cells (reviewed in (Shastri et al., 2013). This body of evidence suggests that C1q may exert a detrimental role in AD by contributing towards the aggregation of fibrillar amyloid, causing an increase in inflammation and an activation of the classical complement cascade. The major consequence of which is a loss of neuronal and neurite integrity.
Seemingly at variance with these compelling studies, many researchers have hypothesised that the role of C1q and the complement system in AD is neuroprotective in nature. Some studies have bolstered this postulation. Foremost were the observations that C1q deficient individuals develop systemic lupus (Botto and Walport., 2002) and APP C1q-/- mice of a particular strain display an SLE-like phenotype, evident in the persistence of glomerulonephritis. The hallmarks of the disorder are multiple apoptotic cell bodies and immune deposits, assessed by immunofluorescence and electron microscopy (Botto et al., 1998). Providing an in-vitro correlate, subsequent experiments have shown that globular head domains of C1q bind to apoptotic cells (Navratil et al., 2001), thus inducing activation of the complement system (Nauta et al., 2002). The stability of the binding between the cells and C1q increases during the latter stages of apoptosis, due to the shedding of complement inhibitors from the cell membrane in early apoptosis (Trouw et al. 2007; Fraser et al. 2010). Moreover, C1q through direct binding and opsonisation of target cells, or by binding IgM on late stage apoptotic cells has been shown to contribute to apoptotic cell clearance (Ogden et al. 2001) (Chen et al. 2009). Thereby leading to the activation of the CCP, recognition by phagocytes such as microglia, and subsequent phagocytic clearance (Trouw et al., 2007). A reduction in inflammation and inflammatory markers was also observed in this study. C1q itself does not induce pro-inflammatory cytokine production. It inhibits the activation of NFkB, an important pro-inflammatory regulator and thus leads to a resolution of inflammation (Fraser et al., 2007). Interestingly, C1q may serve as a reliable marker of microglial activation (CNS resident macrophage), ranging from undetectable levels of C1q biosynthesis in resident microglia to high C1q expression in activated, non-ramified microglia. Microglia added to C1q coated wells or fed apoptotic neurons, or neuronal blebs coated with C1q suppressed the expression of IL- 1α, IL-1β, IL-6 and TNF-α (Fraser et al., 2010).
Wyss-Coray and colleagues reported that “activation of the CCP can attenuate the effects of AB mediated toxicity, and may also reduce the amyloid load either by promoting clearance of senile plaques or reducing their accumulation” (Wyss-Coray et al., 2002). Furthermore, it was previously reported that AD mice expressing the soluble CR1 an inhibitor of complement protein C3 displayed more AB deposition and further progressed neurodegeneration than age-matched controls (Fonseca et al., 2004). Concurrently, complement inhibitor C4b-binding protein (C4BP) was found to be present on apoptotic cells and in AB plaques in-vivo (Trouw et al., 2008). C4BP inhibits the activation of the MAC, opsonisation of cells and phagocytic clearance. Thus, C1q activation and the subsequent formation of the MAC play a decisive role in the induction of neuronal apoptosis and phagocytic clearance in AD (Trouw et al., 2008).
Regulation of the complement system does not solely rely on the expression of the complement proteins. In order to complete it’s full innate immune role it relies on the efficacious binding to its receptor components which are present on many CNS cells. One such example of disruption of normal complement function is the decreased concentration of CR1 (also known as C3b/C4b or CD35 receptor) encoded by the previously described CR1 gene in AD. Due to a gene variant, which produces low copies of the protein, the proportion of C3b opsonised immune complexes that are phagocytosed will be significantly lower than in an age- matched control. As such inflammation will persist in a chronic state, as the toxic species continue to reside in the CNS milieu. C4BP, CD46 CD55 are inhibitors of C3b, which prevent formation of the MAC, with the former residing on the surface of apoptotic membranes thus inhibiting phagocytosis and ensuring the persistence of necrotic cells (Trouw et al., 2008).
Insufficiency or deficiency of complement inhibitors may also play a significant role in the pathogenesis of AD. CD59, which prevents MAC by forming a complex with C5b-8, has been shown to be significantly decreased in the frontal cortex and hippocampus of AD compared with non-demented elderly patients (Yang et al., 2000), whereas C9, the terminal component of MAC, is significantly increased. Aβ-peptide can down-regulate CD59 expression at the mRNA level, suggesting a partial explanation for CD59 deficit in the AD brain (Kolev et al., 2009).
The evidence presented for either a neuroprotective role or a neurodetrimental role for C1q in AD, though seemingly contradictory are equally compelling. Both streams of research implicate C1q as an important regulator of internal homeostasis. We postulate that the different roles observed are due in large part to the presence of fibrillar amyloid and its activation of the complement system. In the absence of fibrillar amyloid we suggest that CCP activation, opsonisation and phagocytic clearance will occur. However, In its presence activation of the CCP will occur, but due to inhibitors of downstream complement factors such as C4BP, Crry, formation of the MAC will not occur. As a result, apoptotic cells will persist within the CNS parenchyma and inflammation will be perpetuated. In order to combat this, two streams of action may be taken. One such option would be to increase the amount of C1q. However, we suggest this would likely have minimal effect, as C1q is already upregulated in AD. Hence, the more likely avenue for treatment would be the creation of recombinant proteins, to provide competitive binding for Aβ, thus reducing CCP activation and the recruitment of microglia to the target sites.
From the experimental evidence shown above it is evident that C1q, the CCP and the MAC play a role in the perpetuation of chronic inflammatory state observed in AD. Whether through direct binding to Aβ and the neuronal membrane, impairment of microglial phagocytosis, the indirect contribution to amyloid aggregation; the detrimental effects of C1q far outweigh its innate immune protection. As such we posit that the CCP and its primary recognition component C1q are viable targets, which when inhibited may have therapeutic implications either as a sole therapy or as a co-adjuvant to currently existing therapies.
In this study we aim to prove the hypothesis by:
- Cloning mutant forms of globular head, which lack the collagen region, in order to identify the impact subtle variations in the protein sequence may have.
- Express the recombinant proteins globular head A, globular head B, globular head C.
- Express recombinant protein mouse variants mghA, mghB and mghC.
- Express the mutant heads lacking the collagen region, which we have, denoted ghA3, ghB3 and ghC3. Examine the proteins to identify what form they take.
- Examine the interaction between the recombinant ghA, ghB, and ghC and Aβ-42.
- Examine the interaction between the mouse ghA, ghB, and ghC and Aβ-42.
- Examine the interaction between ghA3, ghB3 and ghC3 and Aβ-42
- Examine the ability of ghA3, ghB3 and ghC3 to inhibit haemolysis of sheep erythrocytes.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biomedical Science"
Biomedical Science focuses on how cells, organs and systems function in the human body and underpins much of modern medicine. Biomedical Science applies parts of natural and/or formal sciences to help develop advances in healthcare.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: