Cancer Immunotherapy: a Sign of Curative Cancer
Info: 7613 words (30 pages) Dissertation
Published: 9th Dec 2019
Abstract
The advancement of molecular immunology led to the emerging of cancer immunotherapy. The cancer immunotherapy triggers different immune cells to initiate anti-tumor response and subsequently eradicate the tumor. Immunotherapeutic strategies including immune checkpoint blockade, antibody drug conjugate (ADC), oncolytic therapy and particulate system as well as cancer vaccine have shown promising results in several animal models and patients. The clinical successes of anti-CTLA-4 and anti-PD-1 have clearly changed the paradigms of clinical cancer managements. In this review, we discuss some of the today cancer immunotherapy modalities and highlight challenges for the next generation novel immunotherapeutic agents.
Keywords: T cell, Immune checkpoint blockade, Oncolytic virus therapy, anti-tumor response, cancer immunotherapy, Cancer Vaccine, Modalities.
1. Introduction
Cancer is one of the most leading causes of morbidity and mortality in the whole nation [1]. Scientists and clinicians believed that cancer will be the number one cause of death in the United States, elevating the pressure to find an effective alternative therapy [1]. Individual tumor cell appears as result of abnormal changes in the genetics and epigenetics materials leading to cell immortality as well as creating a foreign antigen, referred to as neo-antigens, which theoretically renders neoplastic cell recognizable by the immune system [1]. Despite the capability of the immune cell to detect several abnormal protein structures at the atomic level, cancer cells find their way to escape the immune system recognition, through different mechanisms such as induction of tolerance and disruption of T-cell signaling [1]. Utilizing a mechanism called immune editing, tumor cells will be able to manipulate the immune system to spread more aggressively and become less immunogenic [2].
For decades, the Immune system has been known to be a major player in fighting cancer cells growth. In fact, the full mechanism of interaction between immune and tumor cells was considerably unknown[3].Also, the significant hurdle related to the in vitro culturing of immune cells is one the barriers in advancing targeted cancer immunotherapy [4]. Following large number of unsuccessful trials and failed experiments, the cancer immunotherapy field has received significant attention after the great success of the front runner immunotherapeutic agents in the clinic. Some examples includes the approval of first autologous cellular immunotherapy, sipuleucel-T , for the treatment of prostate cancer in 2010; the approval of the anti-cytotoxic T lymphocyte-associated protein 4 (CTLA-4) antibody, Ipilimumab, and the anti-programmed cell protein 1 (PD1) antibody ,Pembrolizumab, for the treatment of melanoma in 2011 and 2014, respectively (Table 1)[5]. Indeed, Immunotherapeutic agents offer a better and yet a safer novel alternative, in comparison with chemotherapy or radiotherapy, by mounting the immune cells to specifically target the neoplastic cells [5]. Targeted immunotherapy is being currently investigated to combat all kind of cancers[6]. In this review, we discuss the immune cells mounting mechanisms against cancer cells and the current immuontherapeucits treatment modalities. Then, we briefly demonstrate some of the challenges for the cancer immunotherapy
2. Immune cells in Cancer Immunotherapy.
Among the various types of the human body immune cells, the T and B lymphocytes are the major elements in tumor immunity[7]. Th1 CD4+ T cells secrete mainly IFN-γ and IL2 cytokines, which contribute to the activation of macrophages and differentiation of T cell to cytotoxic effector cell ,respectively.[7] On the other hand, Th2 CD4+ T cells secrete different set of cytokines ( e.g. IL4, IL5 ) causing the maturation of B cells to plasma cells, the particular important cells in producing antibodies[7]. The antibodies has two critical roles by either eliminating pathogens directly or assisting other cell, such as natural killer (NK), to kill pathogens in an efficient mechanism called Antibody Dependent Cell Mediated Cytotoxicity (ADCC)[8]. While the CD4+ T cells are MHC (Major Histocompatibility Complex) class II restricted, the cytotoxic T cells CD8+ are MHC class I restricted [7]. The MHC restriction is one of the immune cells mechanisms of Interaction with body cells[7]. Moreover, MHC class I presented in most of the body cells but the MHC class II are certainly expressed in certain macrophages and dendritic cells, named as the antigen presenting cells (APCs) [7]. The maturation of the APCs initiates an antigen presentation process in the secondary lymph nodes followed by Th1 or Th2 activation pathways[7].
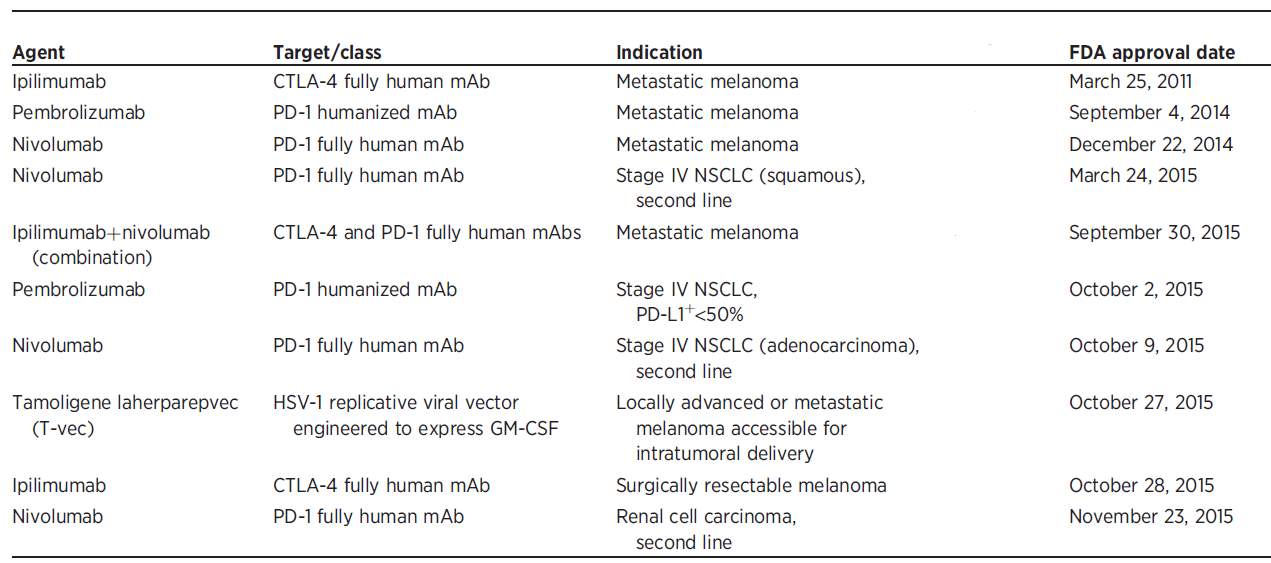
Table 1. Chronological FDA approval of numerous Immunotherapeutic agents. Reprinted with the permission from
3. Types of cancer immunotherapy.
Based on the antitumor mechanisms, cancer immunotherapy is divided to two categories, passive or active.
3.1. Passive immunotherapy
Passive immunotherapy uses a biological molecule, mainly made outside of the body, to attack the cancer cells [9]. Most of the monoclonal antibodies (mAbs) in the clinics today are either bind to specific tumor antigen to inhibit the growth, or initiate apoptosis mechanism by engaging with pro-apoptotic target[9]. Moreover, mAbs help in antigen expression to numerous APCs[9]. Professional antigen presenting cells, another scientific term for DCs, have been found maturated in patients treated with mAbs[9]. Cetuximab is an example of mAb that binds to EGFR receptor and causes DCs maturation.[9] Furthermore, mAbs can activate immune cells component (e.g. Fc portion) attracting NKs cells to destroy the cell attached to the mAb via ADCC mechanism[9].
Cytokines therapy, represents another class of the passive immunotherapy, has been studied to facilitate the immune cells stimulation[10]. IFN-γ and IL-2 are examples of cytokines therapy co-stimulate CD8+ T cells and macrophages, respectively[10]. Despite their success in therapeutic application, cytokines are associated with many side effects due to mainly the un-selectivity of triggering immune reactions in the body[10].
3.2. Active immunotherapy
The main approach to selectively recruit T cells against the cancer cells is the “ Cancer Vaccine “[11]. For decades, vaccine has been proven to be safe and effective in fighting pathogens, such as virus and bacteria[11]. Based on the mechanism of protection, cancer vaccine is classified to prophylactic or therapeutics[11]. The prophylactic vaccines are chiefly composed of a viral protein/s, with or without adjuvant, in which they stimulate the B cells to secrete antibodies specifically bind to the virus and ultimately prevent the viral infection[11]. Cancer vaccine has emerged in cancer therapy field following the great success of Engerix-B® and Gardesil®, against Hepatitis B virus (HBV) and Human Papillomavirus (HPV), respectively[12]. The latter examples have been significantly effective to lower liver and cervical cancer incidences, respectively,[12]. Nevertheless, therapeutic vaccines utilize a new pathway of targeting cancer cells[12]. The vaccines promote APCs maturation and subsequent stimulation of cytotoxic T lymphocytes (CTL) and macrophages [11, 12]. Sipuleucel-T , the first cancer approved therapeutic vaccine, is showing trivial results in treating patients with prostate cancer[12].
4. Cancer immunotherapy modalities
After the discovery of immune checkpoint blockade, there are several novel treatment modalities that differ in their structure, mechanism of action and tumor targeted receptor under development as well as many treatment modalities currently being used in treating cancer patients.
4.1. Monoclonal antibody therapy
The use of the monoclonal antibodies (mAbs) therapy was primarily described by Paul Elrich as magic bullet. However, it hasn’t been introduced to treat patient until the approval Rituximab, the first mAb, in 1997 [13]. The mAbs have been proven to (1) induce tumor cytotoxicity directly , (2) mediate immune cells tumor cell killing and (3) inhibit the uncontrolled vessels and stromal cell growth (Figure 1) [13]. Two major mAbs category is currently in the market, antibody-drug conjugate (ADC) and the Immune checkpoint blockade.
4.1.1. Antibody-drug conjugates (ADC)
The first generation of the mAbs (e.g. Herceptin and Rituximab) has been exploited as efficient receptor signaling inhibitors to block the tumor growth and induce apoptosis[13]. Nevertheless, significant antitumor cytotoxicity have been achieved only at high concentration which led to development of the new generation of maAbs, antibody-drug conjugate (ADC)[14]. Although the concept of drug conjugating confronted major obstacles at the beginning , it became one of the useful immunotherapies in preference for many patients today[14].
ADCs significantly increase the potency of several cancer chemotherapeutic agent[15]. Doxorubicin potency , for example, has increased from 10−7 M to 10−11 M following the conjugation to a mAb[15]. Whereas mAbs cytotoxicity comes directly from engaging to expressed tumor receptors, many ADC cytotoxicity synergized by an additional antitumor mechanism, ranging from interfering with tumor cell DNA signaling to targeting the tumor microenvironment [15]. The addition of auristatins, anti-mitotic agent, to the IgG antibody against CD30 ,Brentuximab, resulted in the development of brentuximab vedotin (or SGN-35), which induces both apoptosis and cell cycle arrest in tumor cell[15]. Brentuximab vedotin showed magnificent results in a phase II clinical trial with a complete remission of 17 refractory systemic anaplastic large cell lymphoma (ALCL) patients and partial remission in 9 others out of 30[15].
Trastuzumab , the FDA-approved IgG1 mAb to target the human epidermal growth factor receptor 2 (HER2) , in conjugation with maytansine derivative( DM1) is another derivative of the new mAbs generation[16]. The trastuzumab-DM1 successfully inhibited the tumor growth of a trastuzumab/lapaninib (a kinase inhibitor used in breast cancer therapy)-resistant mouse model via multiple mechanisms including induction of apoptosis, mitotic catastrophe and ADCC.[16] In phase II clinical trial, trastuzumab-DM1 not only improve the clinical response to the ADC but also significantly lowered the rate of relevant adverse effects ( 37% vs. 75% ) relatively to the standard treatment (trastuzumab plus docetaxel)[16]. Thus, the ADC therapeutic treatments applications has exhibited a trivial success in different clinical trials.
4.1.2. Immune checkpoint blockade.
The body immune response to the neoplastic antigen was observed in many preclinical models and patients to be weak and undetected with the cancer vicinity [3]. After several trials and errors, it was found that cancer cells express inhibitory receptors that shut down many immune cells necessary for activation[3]. One emerging strategy to retrieve the immune cells activity through blocking the inhibitory pathway, termed later as “Checkpoint blockade”[3]. The primary target for checkpoint mAbs is CTLA-4, a receptor expressed in numerous APCs and interferes with CTL activation signaling (Figure 1) [17]. The anti CTLA-4 mAbs restored the CTLs activity and revealed tumor cytotoxicity in many murine tumor models, such as ovary, bladder, and brain[17]. For example, Ipilimumab on phase III trials enhanced significantly the overall survival in patients with metastatic melanoma diseases[17]. The idea of activating T cell component revolutionizes the field of cancer immunotherapy[17].
PD-1 receptor is another inhibitory receptor expressed by CTL and subsequently leads to inhibition of T cell proliferation, cytotoxicity and cytokine release [18, 19]. Following the interaction of PD-1 to tumor PD ligands (e.g. PD-L1 or PD-L2), it initiates specific signals that suppress the antitumor activity[18, 19]. Two anti-PD1 mAbs ,Pembrolizumab and Nivolumab , have recently gained FDA approval treatment of metastatic melanoma and NSLC, respectively(Table 1)[18, 19]. Managed by steroid treatment ,PD-1 mAbs have a favorable toxic profile over the CTLA-4[18, 19]. Further, Chen et al. reported massive T- cells activation in the CTLA-4 knockout animal model as opposite to the delay onset of organ specific inflammation. Cleary, the latter evidence emphasize on the importance of tumor targeted selectivity to reduce the toxicity related to immune cell mounting [20].
The complexity of the cancer immune inhibitory mechanisms is the main focus of scientists nowadays, and new inhibitory receptors are being discovered in a regular daily basis[21]. Lymphocyte activation gene 3 (LAG3) and mucin domain-containing 3 (TIM3) protein are two of many molecules under clinical development and being tested in clinical trials [21].
4.2. Oncolytic virus therapy.
The discovery of immune suppressive tumor environment on one hand improves the tumor cell survival but on other hand limited their capability to respond to viral infection[22, 23]. The observed theory of “ hallmarks of cancer” including sustained proliferation, usurping cellular apoptotic programs, and inactivating growth suppressors, represents the ideal environment for selective replications of viruses[22, 23]. The knowledge of this theory led to development of oncolytic virus (OV)[22, 23]. OVs have three distinctive mechanism of tumor killing (1) acute tumor debulking owing to tumor cell infection and lysis (2) trigger vascular disturbing effect and (3) elicit antitumor immunity (Figure 2).[22, 23]
OVs genetic material manipulation opens many targeting opportunities to augment the antitumor activity and minimize the unspecific pathogenicity[24]. One of the approaches is to induce deletion of pathogenic genes to restrict the growth in cancer cells rather than healthy normal cells[24]. Also, OVs gene could be modified to express certain cytokine and hence recruits numerous T cells to kill cancer cells[24].
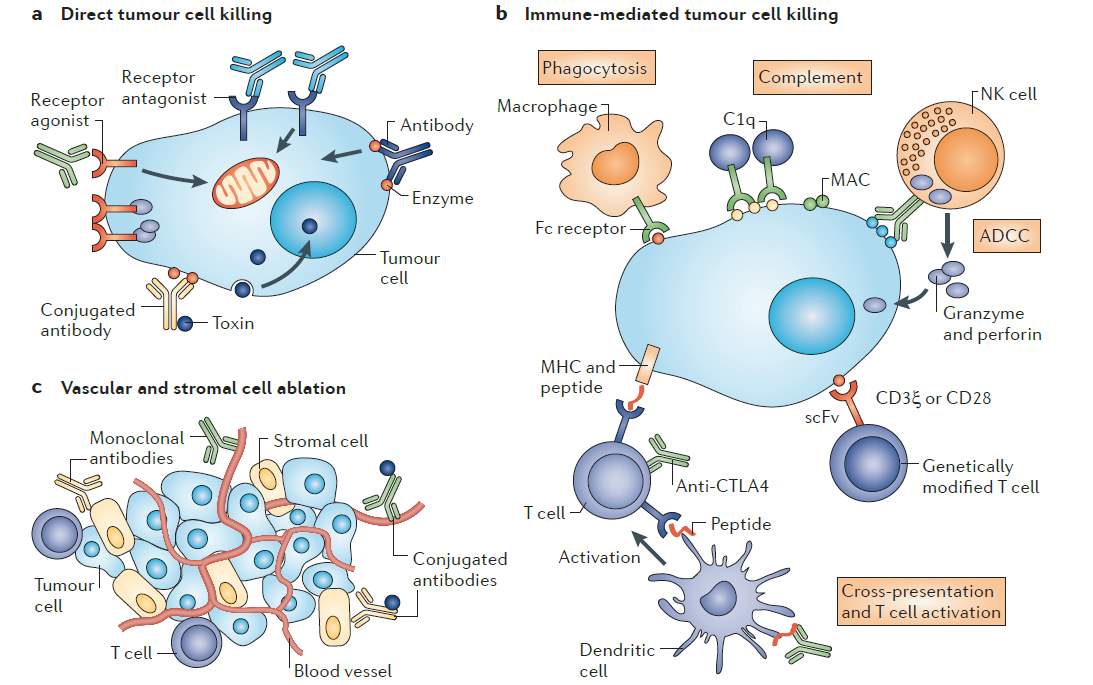
Figure 1. Monoclonal antibody antitumor mechanism. (a) Direct tumor cell killing could be initiated by receptor agonist, such as mAb binds to a tumor cell receptor inducing apoptosis. Alternative mechanism is via delivering cytotoxic molecules to directly cause cell lysis or interfere with essential survival pathway for tumor cells, antibody-drug conjugate (ADC). (b) Immune-mediated tumor cell killing can be elicited by different immune cells including macrophages, NK cells and T- cells CD8+ activated cells. Particularly, immune checkpoint blockade mAbs; Anti-CTLA-4 antibodies bind to T-cells receptor to restore T- cells antitumor activity. (c)Vascular and stromal cell ablation is generated mainly through targeting abnormal vascular system by antibody-drug conjugated (ADC). It also could be induced by blocking a survival receptor by mAbs. NK, natural killer; CTLA4, cytotoxic T lymphocyte-associated antigen; MHC, Major histocompatibility complex. Reproduced with the permission from reference
After the infected tumor cells lysis, the tumor antigen released is taken up by APCs, mounting specific and targeted CTL response against the tumor [25, 26]. However, OVs lytic action might not appear at the initial stage of treatment due to the rapid clearance from host immune response [25, 26]. PEGylation (conjugating the viral coat with polyethylene glycol) is one of the techniques used to overcome such drawback and block the neutralizing antibodies response [25, 26]. Other techniques, such as inhibit host immune response by pretreatment with cyclophosphamide or block the viral gene expression to diminish the antigen presentation, are well-established for OVs therapy [25, 26].
Different type of viruses for OV therapy has been evaluated including Newcastle disease virus (paramyxovirus), reovirus, measles virus (paramyxovirus), and vaccinia virus (poxvirus)[27]. The most advanced OV agents reached out to the clinic is Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma[28]. The T-VEC is a herpes simplex virus with genetic deletion of two ICP34.5 genes to eliminate neuronal side effects[29]. Additionally, the virus gene was grafted with GM-CSF cytokine gene sequence in an effort to recruit and differentiate DCs within the tumor vicinity and subsequently improve the antitumor activity[29].
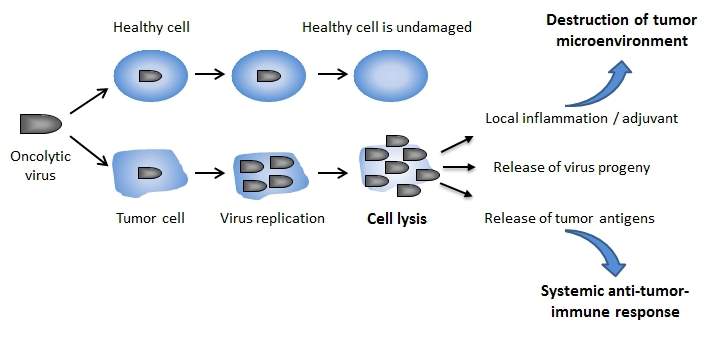
Figure 2. Oncolytic virus mechanism of actions. Oncolytic virus (OV) are engineered to selectively infect tumor cells and cause minimal damage to healthy cells. Consequently, OV exhibits a series of antitumor cytotoxicity effects primarily by causing cell lysis, disturbing the vascular in tumor microenvironment (not shown) and recruiting immune cell as result of tumor antigen release. Reproduced with permission from reference
T-VEC therapeutic outcomes from preclinical studies in many animal models and tumor cell lines were very promising.[30] A phase II clinical trial was carried out and study investigators reported an objective response rate of 26 % with flu-like symptom in advanced melanoma patients[30]. Theose encouraging results were positively translated 439 nonresectable melanoma (stages IIIB, IIIC, or IV) enrolled in multi-central phase III clinical trial[30]. The primary end point was durability response rate for subjects randomized 2:1 to T-VEC or GMCSF[30]. The durability response rate for T-VEC was (16.3 vs. 2.1; P < 0.001) compared to control arm in the study[30]. Finally, most of the patient experienced fatigue, fever and nauseas as well as irritation at the injection site[30]. Based on the previous and other findings, the T-VEC was the first OV immunotherapy gets the FDA approval in October 2015(Table 1)[28].
Although T-VEC therapy has been in the market for two years, the virus integration to the clinic has been very slow[31]. In a recent clinical review of T-VEC, the author explained the OV limitations as uneducated health care providers, unprepared storage conditions for viruses in hospitals and unfamiliar T-VEC administration since it has to be given intratumorally with assist of ultrasound device or clinical palpation[31].
4.3. Particulate system therapy.
The advancement of nanotechnology has obtained a great deal of attention in medical applications[32]. The proof-of-concept was very encouraging after the FDA approval of Abraxane® (paclitaxel nanoparticles) and Doxil® (doxorubicin liposomes)[32]. Many particulate systems have sought a bright future in the application of cancer vaccine as they facilitate antigen release in controlled manner overcoming the short-lived release of subunit vaccine[33, 34]. Furthermore, the particulate system presented with interesting properties including antigen presentations, long-time antigen release, multivalent antigens and targeting[33, 34]. Shen et al. tested antigen uptake and CD8+ T-cell activation in culture with DCs pretreated with encapsulated polymeric nanoparticles, poly(lactic-co-glycolic acid) (PLGA), or soluble antigen[33, 34]. The T-cell response achieved in the nanoparticle study group was
at 1000-fold lower antigen concentration than the soluble antigen[33, 34].
Several particles have been explored in cancer vaccine, majority of which are polymeric nanoparticles, metallic nanoparticles, and polymeric micelles[32]. However, scientists prefer biologically derived particles due to the low immunogenicity and excellent biocompatibility[32]. The most attractive candidates are virus-like particles, non-viral protein self-assembled nanoparticles and lipid-based particles [32].
4.3.1 Virus-Like particles
Virus-like particles (VLP) are multimeric nanoparticles assemble from viral proteins and lack of viral genome [35]. Based on the cell membrane structure, VLPs are classified to enveloped and non-enveloped viruses[36]. Enveloped VLPs play a keystone role in antigen presentation due to the fact that full-length monomeric or multimeric protein could be anchored through transmembrane domains[36].
VLPs are very efficient particles in delivering antigen to APCs for three critical reasons (1) The VLPs well-fit size for lymphatic draining and hence antigen delivery to a rich region of APCs (2)The VLPs shape that mimics the structure of virus with multiple conformational epitopes in the outer membrane (3)The VLP has a good tolerability as result of APCs specific targeting [36]. For example, the immune potentiators are encapsulated and their non-specific serum protein binding significantly diminished[37]. This is a very crucial aspect in delivering immune potentiators molecules[37]. Many systemic side effects have been observed from nonmethylated CpG motifs and other immune potentiators[37]. To date, there are only two VLPs based licensed vaccine, recombinant hepatitis B virus (HBV) and human papillomavirus virus (HPV) vaccine[38].
In 2015, 900 000 death reported from hepatitis and its related complications including cirrhosis and hepatocellular carcinoma caused primarily by hepatitis B virus[38]. The HBV is the smallest DNA virus from the Hepadnaviridae family[39]. A 40 nm double-shelled virion was observed in the virus from infected human serum[39]. The outer envelope is composed of lipid and three proteins, in which they form a self-assemble sub-viral particles called ”HBsAg”[39]. Additionally, It was noticed that HBsAg sub-viral particles in 103 to 105-fold excess over the integral virion particles[39].
During the vaccine production infancy, the infected sub-viral particle were purified and used for immunization in a dose of 20 μg/ml sub-virion particles of HBsAg[40]. Nevertheless, the safety of the vaccine and availability of donor’s serum were major obstacles for the vaccine production[40]. The advancement of DNA recombinant technology led to the development of new vaccine generation that utilizes eukaryote expression hosts such as yeas and mammalian cells[40]. The vaccine has dramatic reduction in the number of hepatocellular carcinoma cases since its introduction[41] For instance, Taiwan was a high epidemic area of the hepatitis B with 20% of the adult population were considered to be HBV carrier [41]. After 30 years of the vaccine implantation, the prevalence of HBsAG among children younger than 15 has significantly decreased from 9.8 % to 0.9 % [41].
Human papillomavirus (HPV) belongs to Papillomaviridae family and it is the causative agent for many warts in the human mucosa and skin[42]. Papillomavirus has been known for decades to cause cervical cancer, the second most common cause of women death globally[42]. Countless types of papillomavirus have been recognized with 15 virus types linked to cervical cancer (HPV-16, -18, -31, -33, -35, -45, -52 and -58) [42]. Evidently, HPV type 16 is one order magnitude higher than other types in causing cervical cancer [42]. The HPV is made of two distinguishable encoded protein identified as L1 and L2[43]. The L1 is the most abundant form and self-assemble to 60 nm particles[43]. In animal models, the L1 particles generate significant level of antibodies beside protect the animal from later challenge with the virus[43].
Gardasil® vaccine from Merck and Co., Inc. vaccine obtained the first FDA approval in 2006[42]. Its quadrivalent (HPV- types 6, 11, 16 and 18) VLP-based vaccine produced via introducing L1 gene in a yeast, Saccharomyces cerevisiae[44]. Whereas HPV type 12 and 16 are high risk associated with cervical cancer, type 6 and 11 are associated genital warts[44].1 The second HPV vaccine is Cervarix® , which was approved by the FDA in 2009 (Table 3)[44]. Unlike the Gardasil, the Cervarix is bivalent (HPV- 16 and -18) and it is manufactured by GlaxoSmithKline[44]. It’s also expressed in insect, Trichoplusia ni , instead of the yeast as in the case of Gardasil[44]. In a recent systemic review, the infection rate from high-risk of HPV (6, 11, 16 and 18) was documented to be less than 90% in some area [45].

Table 3. Detailed description of licensed human papillomavirus vaccine.
Both of the approved VLP-based vaccines are breakthrough for cancer vaccines[44]. They have been significantly effective in preventing cancer incidence as it’s implanted in universal vaccination programs recommended by WHO and other health organizations[44]. The state-of-arts in the VLP-based vaccines holds a promising future for other particulate systems to be seen in the markets soon.
4.3.2. Non-viral protein self-assembly particles.
In addition to the VLP, there are many non-viral proteins self-assembled particles that show potential applications in cancer immunotherapy[32]. Protein based nanoparticles demonstrates excellent biocompatibility and biodegradability[32]. As discussed early, the first therapeutic cancer vaccine was Provenge®(sipuleucel-T) for the treatment of advanced prostate cancer[12]. Sipuleucel-T is a protein based therapy used to pulse the DCs ex vivo to express prostate cancer antigen[12].
Ferritin is the main cellular self-assembled protein for iron storage and transportations[46]. Ferritin can be assembled to nanoparticles that have great capacity to carry antigens to APCs[46]. Bo et al. studied the lymph node targeting efficiency of four different nanoparticles[47]. The lymphatic nodes (LN) were targeted with Escherichia coli DNA binding protein (DPS), Thermoplasma acidophilum proteasome (PTS), hepatitis B virus capsid (HBVC), or human ferritin heavy chain particle (hFTN)(Figure 3)[47]. hFTN nanoparticles was delivered to LN in less than 1 minute of the incubations with DCs and prolonged the release over 6 days[47]. The author reported a strong attraction between hFTN and T lymphocytes[47]. In the LN region, lymphocytes (B and T cells) express T cell immunoglobulin and mucin domain-2 (TIM-2) which explains the reason of the hFTN particles accumulations for a significant long time[47]. Further, the hFTN protein nanoaprticles were able to inhibit tumor growth of mice challenged with B16-F10 melanoma tumor cells[47]. This study reveals to the feasibility of ferritin nanoparticles in the application of cancer vaccine[47].
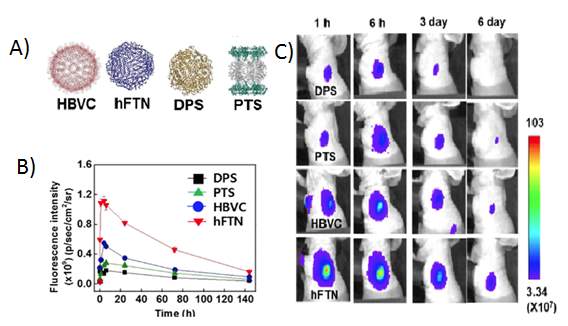
Figure 3. Evaluation of lymph node targeting using different protein nanoparticles.(A) Four different candidates, hepatitis B virus capsid (HBVC), human ferritin heavy chain particle (hFTN), Escherichia coli DNA binding protein (DPS) and Escherichia coli DNA binding protein (DPS). (B) Analysis of the fluorescent intensity of the protein nanoparticle. (C) Fluorescent images of the four study groups at time point 1 h, 6 h, 3 days and 6 days. The entire fluorescent image captures and analysis were determined using IVIS spectrum imaging system. Reproduced with the permission form reference.
In another attempt for cancer vaccine delivery, non-vial E2 subunit of pyruvate dehydrogenase particulate system has been investigated [48, 49]. E2 protein assembled to 25 nm dodecahedral nanoparticles with high structure stability [48, 49]. In a new study, Nicholas et al. designed E2 protein based tumor vaccine, referred to as viral-mimking protein nanoparticles, to target DCs and other APCs [48, 49]. The CpG motif was encapsulated in the nanoparticle while the MHC I-restricted SIINFEKL peptide epitopes were expressed on the surface [48, 49]. The study findings proved that the nanoparticle elicited a significant presentation by DC relative to unbound form of the individual components, the CpG and the peptide [48, 49]. In a different study, the previous authors further used the E2 protein nanoparticle expressed with a new epitope in animal models [48, 49]. Tumor associated antigen, Gp 100 melanocyte differentiation protein (gp 100), was chemically conjugated to the particle surface[48, 49]. After single immunization with the nanoparticles, the frequencies of epitope-specific CD8+ T-cells were 30-fold and 120-fold higher in spleen and lymph nodes, respectively [48, 49]. Interestingly, the nanoparticles promotes an increase of 40% in animal survival time, compared to the control group [48, 49]. The two previous studies highlight on the significance of mimicking virus structures to achieve significant DCs antigen uptake as well as CD8+ activation.
4.3.3. Lipid based particles.
Lipid based particles have a similar structure to the cell membrane, an ideal property to deliver numerous cargos inside cells [50]. Most of the vesicles used to transport cellular cargo within and outside the cells, such as microvesicles and exosomes, are lipid based vesicles [51]. In fact, exosomes plays a critical role in communicating between immune and tumor cells [51]. Exosomes are small lipid vesicle in the size of 40 of 100 nm consisting of a mix of lipids and proteins (Figure 4) [51]. Three types of exosomes have been described, ascitic cell-derived exosomes (AEX), dendritic cell-derived exosomes (DEX) and tumor cell-derived exosomes (TEX) [51].
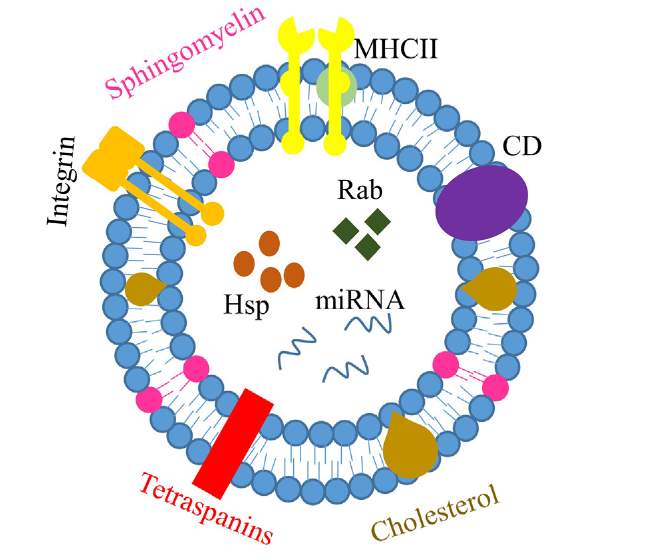
Figure 4. Main compositions of exosome. Exosomes are basically small bilayer vesicles with proteins and lipids. It contains a variety of protein including integrin, cluster of differentiation (CD), and Major Histocompatibility complex (MHC II). Also, it has certain types of lipids, cholesterol and sphingomyelin. Nucleic acid is encapsulated in the enternal compartment. Reproduced with the permission from reference.
AEXs purified from peritoneal cancer patients were able to stimulate DCs presentation and subsequently induce CD8+ T cell activation pathways [52]. Besides inducing CD8+ T-cells simulations, TEXs have been confirmed to mount a Th1 CD4+ T cell response due to the fact that TEXs express heat shock protein (Hsp 70)[53]. However, DEXs seem to have the highest effect in term of antigen presentation and T cells activation [54]. In a side by side study, the TEXs were compared to DEXs in DCs pulsing with ovalbumin (OVA) as a model antigen [55]. Unlike the TEXs, the DEX expressed co-stimulatory molecules, CD40 and CD80, which exhibited a potent immune response and CTL activation [55]. Other studies reported an involvement of different immune cells, particularly Natural kill (NK) cells, to inhibit tumor cell growth [55]. In phase 1 clinical trial for DEXs in non-small cell lung cancer patients, DEXs loaded with tumor antigen were well-tolerated and showed a great feasibility for large scale production [56]. Although there was inconsistency in the immune antigen specific T cell activation, NK effector cells were detected in majority of the patients enrolled in the study [56]. To date, scientists are still interesting to understand the communication dynamic between immune and tumor cell [51]. Thus, it could assist to generate more immunogenic exsosomes for cancer vaccine applications.
5. Limitations and challenges.
Current promising evidences of different immunotherapy modalities are behind the motivation of the immuno-oncology field in academia and industry [57]. Several investors and health care analysts predict a market size of US$35 billion by 2023 [58]. Nevertheless, toxicity is a potential barrier for the immunotherpay applications[57]. Whereas several immunotherapy studies demonstrate a bright site of manageable adverse events, the dark side of immunotherapy could be a tragic [57]. Disturbing the immune balance between immune cells and tumor could lead to overgrowth of tumor cells [57]. For instance, the re-activated immune system might generate more immunosuppressive cytokines that proliferates tumor cells and cause metastasis [57]. Additionally, T regulatory cells (T reg) response will be activated via “Rebound Effect” causing expansion of regulatory key elements to diminish the antitumor effect [57]. The latter barrier and other unexplained activation mechanism have to be carefully addressed for the next generation immunotherapeutic modalities.
6. Conclusion and Future prospective.
Cancer therapy has been the focus for the scientists for decade to develop a new effective therapy and lower the mortality rate from cancer. Cancer immunotherapy and vaccines is nowadays an excellent treatment option for many tumor resistance patients. Immune checkpoint blockade has been the breakthrough in the immuno-onclogy field. In fact, James P. Allison was awarded with Lasker-DeBakey Clinical Medical Research Award for the development of the first immune checkpoint blockade, Anti-CTLA-4. The clinical success is truly translated by the number of FDA approved immune checkpoint blockades mAbs .The mAbs approval opens new avenues for other treatment modalities to recruit immune cells and eventually fight against cancer cells. Cancer immunotherapy may pave the way toward curative cancer.
References
1. Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med 14 73 (2016).
2. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 14(8), 561-584 (2015).
3. Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. Journal of Clinical Oncology 29(36), 4828-4836 (2011).
4. Snook AE, Waldman SA. Advances in cancer immunotherapy. Discovery medicine 15(81), 120 (2013).
5. Gu L, Mooney DJ. Biomaterials and emerging anticancer therapeutics: engineering the microenvironment. Nat Rev Cancer 16(1), 56-66 (2016).
6. Brodsky FM, Guagliardi LE. The cell biology of antigen processing and presentation. Annual review of immunology 9(1), 707-744 (1991).
7. Luckheeram RV, Zhou R, Verma AD, Xia B. CD4< sup. Clinical and developmental immunology 2012 (2012).
8. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature medicine 10(9), 909-915 (2004).
9. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nature reviews cancer 12(4), 237-251 (2012).
10. Baluna R, Vitetta ES. Vascular leak syndrome: a side effect of immunotherapy. Immunopharmacology 37(2), 117-132 (1997).
11. Yaddanapudi K, Mitchell RA, Eaton JW. Cancer vaccines: Looking to the future. Oncoimmunology 2(3), e23403 (2013).
12. Ramqvist T, Andreasson K, Dalianis T. Vaccination, immune and gene therapy based on virus-like particles against viral infections and cancer. Expert Opin Biol Ther 7(7), 997-1007 (2007).
13. Peters C, Brown S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep 35(4), (2015).
14. Fuenmayor J, Montaño RF. Novel Antibody-Based Proteins for Cancer Immunotherapy. Cancers 3(3), 3370-3393 (2011).
15. Alley SC, Okeley NM, Senter PD. Antibody–drug conjugates: targeted drug delivery for cancer. Current opinion in chemical biology 14(4), 529-537 (2010).
16. Perez E, Dirix L, Kocsis J et al. Efficacy and safety of trastuzumab-DM1 versus trastuzumab plus docetaxel in HER2-positive metastatic breast cancer patients with no prior chemotherapy for metastatic disease: preliminary results of a randomized, multicenter, open-label phase 2 study (TDM4450G). Presented at: Annals of Oncology. 2010.
17. Ribas A. Releasing the brakes on cancer immunotherapy. N Engl J Med 373(16), 1490-1492 (2015).
18. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. The EMBO journal 11(11), 3887 (1992).
19. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26 677-704 (2008).
20. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 39(1), 1-10 (2013).
21. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. Journal of Experimental Medicine jem. 20100643 (2010).
22. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. cell 144(5), 646-674 (2011).
23. Russell SJ, Peng K-W, Bell JC. Oncolytic virotherapy. Nature biotechnology 30(7), 658-670 (2012).
24. Hu JC, Coffin RS, Davis CJ et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clinical cancer research 12(22), 6737-6747 (2006).
25. Fulci G, Breymann L, Gianni D et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proceedings of the National Academy of Sciences 103(34), 12873-12878 (2006).
26. Tesfay MZ, Kirk AC, Hadac EM et al. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. Journal of virology 87(7), 3752-3759 (2013).
27. Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer immunology research 2(4), 295-300 (2014).
28. Ledford H. Cancer-fighting viruses near market: anticipated approval in Europe and the United States could spur a promising field with a chequered past. Nature 526(7575), 622-624 (2015).
29. Hercus TR, Thomas D, Guthridge MA et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood 114(7), 1289-1298 (2009).
30. Kaufman HL, Bines SD. OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future oncology 6(6), 941-949 (2010).
31. Rehman H, Silk AW, Kane MP, Kaufman HL. Into the clinic: talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. Journal for immunotherapy of cancer 4(1), 53 (2016).
32. Tan A, De La Peña H, Seifalian AM. The application of exosomes as a nanoscale cancer vaccine. International journal of nanomedicine 5 889 (2010).
33. Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity 33(4), 492-503 (2010).
34. Shen H, Ackerman AL, Cody V et al. Enhanced and prolonged cross‐presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 117(1), 78-88 (2006).
35. Noad R, Roy P. Virus-like particles as immunogens. Trends in microbiology 11(9), 438-444 (2003).
36. Ungaro F, Conte C, Quaglia F, Tornesello ML, Buonaguro FM, Buonaguro L. VLPs and particle strategies for cancer vaccines. Expert review of vaccines 12(10), 1173-1193 (2013).
37. Storni T, Ruedl C, Schwarz K, Schwendener RA, Renner WA, Bachmann MF. Nonmethylated CG motifs packaged into virus-like particles induce protective cytotoxic T cell responses in the absence of systemic side effects. The Journal of Immunology 172(3), 1777-1785 (2004).
38. Kane MA, Brooks A. New immunization initiatives and progress toward the global control of hepatitis B. Current opinion in infectious diseases 15(5), 465-469 (2002).
39. Franco E, Bagnato B, Marino MG, Meleleo C, Serino L, Zaratti L. Hepatitis B: Epidemiology and prevention in developing countries. World journal of hepatology 4(3), 74 (2012).
40. Michel M-L, Tiollais P. Hepatitis B vaccines: protective efficacy and therapeutic potential. Pathologie Biologie 58(4), 288-295 (2010).
41. Meireles LC, Marinho RT, Van Damme P. Three decades of hepatitis B control with vaccination. World journal of hepatology 7(18), 2127 (2015).
42. Villa LL. Prophylactic HPV vaccines: reducing the burden of HPV-related diseases. Vaccine 24 S23-S28 (2006).
43. Chen XS, Garcea RL, Goldberg I, Casini G, Harrison SC. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Molecular cell 5(3), 557-567 (2000).
44. Roldão A, Mellado MCM, Castilho LR, Carrondo MJ, Alves PM. Virus-like particles in vaccine development. Expert review of vaccines 9(10), 1149-1176 (2010).
45. Garland SM, Kjaer SK, Muñoz N et al. Impact and effectiveness of the quadrivalent human papillomavirus vaccine: a systematic review of 10 years of real-world experience. Reviews of Infectious Diseases 63(4), 519-527 (2016).
46. Cho KJ, Shin HJ, Lee J-H et al. The crystal structure of ferritin from Helicobacter pylori reveals unusual conformational changes for iron uptake. Journal of molecular biology 390(1), 83-98 (2009).
47. Lee B-R, Ko HK, Ryu JH et al. Engineered human ferritin nanoparticles for direct delivery of tumor antigens to lymph node and cancer immunotherapy. Scientific reports 6 35182 (2016).
48. Molino NM, Anderson AK, Nelson EL, Wang S-W. Biomimetic protein nanoparticles facilitate enhanced dendritic cell activation and cross-presentation. ACS nano 7(11), 9743-9752 (2013).
49. Molino NM, Neek M, Tucker JA, Nelson EL, Wang S-W. Viral-mimicking protein nanoparticle vaccine for eliciting anti-tumor responses. Biomaterials 86 83-91 (2016).
50. Felice B, Prabhakaran MP, Rodríguez AP, Ramakrishna S. Drug delivery vehicles on a nano-engineering perspective. Materials Science and Engineering: C 41 178-195 (2014).
51. Kooijmans SA, Vader P, Van Dommelen SM, Van Solinge WW, Schiffelers RM. Exosome mimetics: a novel class of drug delivery systems. International journal of nanomedicine 7 1525 (2012).
52. Andre F, Schartz NE, Movassagh M et al. Malignant effusions and immunogenic tumour-derived exosomes. The Lancet 360(9329), 295-305 (2002).
53. Xie Y, Bai O, Zhang H et al. Membrane‐bound HSP70‐engineered myeloma cell‐derived exosomes stimulate more efficient CD8+ CTL‐and NK‐mediated antitumour immunity than exosomes released from heat‐shocked tumour cells expressing cytoplasmic HSP70. Journal of cellular and molecular medicine 14(11), 2655-2666 (2010).
54. André F, Chaput N, Schartz NE et al. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. The Journal of Immunology 172(4), 2126-2136 (2004).
55. Pitt JM, Charrier M, Viaud S et al. Dendritic cell–derived exosomes as immunotherapies in the fight against cancer. The Journal of Immunology 193(3), 1006-1011 (2014).
56. Morse MA, Garst J, Osada T et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. Journal of translational medicine 3(1), 9 (2005).
57. Whiteside TL, Demaria S, Rodriguez-Ruiz ME, Zarour HM, Melero I. Emerging opportunities and challenges in cancer immunotherapy. (2016).
58. Hoos A. Development of immuno-oncology drugs [mdash] from CTLA4 to PD1 to the next generations. Nature reviews Drug discovery 15(4), 235-247 (2016).
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Cancer"
Cancer is a disease in which cells grow or reproduce abnormally or uncontrollably. Cancerous cells have the potential to spread to other areas of the body in a process called metastasis.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: