Kinetic Study of Chalcopyrite Leaching with Iron (III) chloride in Methane-sulfonic Acid
Info: 10036 words (40 pages) Dissertation
Published: 10th Dec 2019
Tagged: Chemistry
Kinetic study of chalcopyrite leaching with iron (III) chloride in methane-sulfonic acid
The increasing rigour of environmental standards for copper extraction and the problems associated with the most common lixiviants used in chalcopyrite leaching make necessary to investigate solutions with less environmental impact. The applicability of the methane-sulfonic acid (MSA) as a copper lixiviant with ferric chloride was developed in this study through the analysis of leaching kinetics and characterisation of solid residues. The effects of different variables including temperature, initial acidity, ferric ions concentrations and particle size were determined, a strong dependence of temperature and particle size was found whereas acid and ferric concentrations had a minimum effect over the leaching rate. Even though sulphur layer formation was evident in the final solid the reaction mechanism stablished was a shrinking core model by surface chemical reaction control for spherical particles, implying that the pass through the sulphur layer was not the reaction rate controller. The apparent activation energy for this reaction had a value of 101 kJ/mol calculated by Arrhenius and ‘time to a given fraction’ methods.
Keywords: Chalcopyrite, Leaching, Copper, Ferric chloride, Methane-sulfonic acid (MSA), Hydrometallurgy
- Introduction
Copper has a vital role in the development of our society, its production and commercialisation prevail to date. The increasing industrial application of this metal requires the exploitation of more refractory minerals and low grade ores to supply the demand that is growing in an exponential rate. Hydrometallurgical methods of copper extraction have become the principal practice to produce this type of mineralization being heap leaching the most common but with In situ leaching rising as a profitable alternative for difficult accessed and deep ore bodies (Northey, Haque et al. 2013). Chalcopyrite (CuFeS2) is the most common copper-bearing mineral on earth and represent more than half of the world copper mineral reserves know to date just as is also the most refractory to hydrometallurgical processing (Wang 2005). Due to its nature chalcopyrite requires the addition of an oxidant and a lixiviant paired to perform the oxidation and dissolution of the chalcopyrite, several lixiviants and oxidants have been tested to obtain the fastest copper recovery rate and avoid side precipitations and formation of passivation layers. The most common oxidant tested were ferric (Fe+3) ions combined with different type of lixiviants including HCl, H2SO4 and HNO3 under a wide range of temperatures from atmospheric leaching to high temperatures, and pH under 2.5 (Dutrizac 1990, Hackl, Dreisinger et al. 1995, Havlík, Škrobian et al. 1995, El Meray, Elamari et al. 2000, Hiroyoshi, Miki et al. 2001, Al-Harahsheh, Kingman et al. 2006, Al-Harahsheh and Kingman 2007, McDonald and Muir 2007, Córdoba, Muñoz et al. 2008, Córdoba, Muñoz et al. 2008, Nazari and Asselin 2009, Olvera, Rebolledo et al. 2015, Shiers, Collinson et al. 2016).
Ferric oxidant have proved to be the most efficient oxidant for leaching chalcopyrite under oxidizing conditions in the presence of chloride, several kinetical studies were performed looking to identify the response under specific conditions with industrial implementation porpoises (Table 1), seems to be a consensus on the determination of the reaction mechanism for ferric chloride identified as chemical reaction control in spite of the formation of a surface coating layer, however, the calculation of activation energies is still subject of discussion, the ranges of values obtained are very varied and went from 15 kJ/mol to 93 kJ/mol, tis value is very dependent on the specific characteristics of the sample including the particle size, gangue content, nature and origin (Dutrizac 1981), but also highly dependent on the selection of the empirical rate. For this reason, it is necessary to use different methods of calculation in order to corroborate the value independently of the empirical equation.
Table 1
Literature review of Chalcopyrite leaching with Ferric ions
| T (ᵒC) | Iron (III) [] (M) |
Acid
[] |
Particle size (µm) |
Leach media | Ea
(kJ/mol) |
Mechanism | reference |
| 70-100 | 1 | 1 | -150+105 | Iron (III) chloride in hydrochloric acid | 92.8 | Surface chemical reaction control | (Rath, Paramguru et al. 1988) |
| 45-100 | 0.2 | 0.3 | -20+14 | Iron (III) chloride in hydrochloric acid | 42 | Surface chemical reaction control | (Dutrizac 1981) |
| 3.5-80 | 0.5 – 1 | 0.25-1 | -315+200 | Iron (III) chloride in hydrochloric acid | 55 | Surface chemical reaction control | (Havlík, Škrobian et al. 1995) |
| 23-40 | 0.28 | 0.25 | Disk electrode specimen | Iron (III) chloride in hydrochloric acid | 86.4 | (Orth and Liddell 1990) | |
| 52-85 | 0.1 | 0.2 | Disk electrode specimen | Iron (III) chloride in hydrochloric acid | 69 | Electrochemical kinetics | (Hirato, Kinoshita et al. 1986) |
| 30-90 | 0.2 | -104 +74 | Iron (III) chloride in hydrochloric acid | 15 – 28 | Surface chemical reaction initial stages and diffusion through a product layer later stages | (Saxena and Mandre 1992) | |
| 75-96 | 0.1 | 1 | -37+44 | Iron (III) chloride in hydrochloric acid | 83 | Surface chemical reaction control | (Palmer, Nebo et al. 1981) |
| 55-94 | 0.1 | 0.2 | Disk electrode specimen | Iron (III) chloride in hydrochloric acid | 69 | Surface chemical reaction control | (Majima, Awakura et al. 1985) |
| 55-85 | 0.004 | 5 | 38<x<75 | Iron (III) sulphate in sulfuric acid | 22(< 10 h) 73 (<10 h) | Diffusion from the interface to the bulk solution and Surface chemical reaction control | (Kaplun, Li et al. 2011) |
| 52-90 | 0.25 | 0.5 | <38 | Iron (III) sulphate in sulfuric acid | 79.5 | Surface chemical reaction control for monosized particles | (Al-Harahsheh, Kingman et al. 2005) |
| 45-100 | 0.2 | 0.3 | -20+14 | Iron (III) sulphate in sulfuric acid | 75 | Diffusion through a product layer | (Dutrizac 1981) |
| 40-85 | 1 | 0.2 | Disk electrode specimen | Iron (III) sulphate in sulfuric acid | 76.8 – 87.7 | First stage transport control, second stage chemical reaction control | (Hirato, Majima et al. 1987) |
| 35-68 | 0.08 | pH 1.8 | 70 | Iron (III) sulphate in sulfuric acid | 130.7 | Surface chemical reaction control | (Córdoba, Muñoz et al. 2008) |
| 60-90 | 0.25 – 0.03 | 1 – 0.01 | 12 and 4 | Iron (III) sulphate in sulfuric acid | 83.7 | Electron transport through a product layer | (Munoz, Miller et al. 1979) |
Due to the increasing environmental controls applied to conventional pyrometallurgical techniques associated with H2S production and the prospect of increased exploitation of otherwise non-profitable ores, there is an increase interest on ISL applications. The biggest concern of in situ mining resides in the risk of water and soil contamination from the leaching solution and produced liquors (Seredkin, Zabolotsky et al. 2016), to this fact the selection of a lixiviant with low repercussions in the subsurface is imperative. For ferric ions systems the presence of a strong acid accelerates the dissolution of the chalcopyrite (Lu, Jeffrey et al. 2000, Wang 2005, Lundstrom, Aromaa et al. 2008), which is why the majority studies have chosen to use hydrochloric acid as a lixiviant, however, HCl is well known as one of the most corrosive, and difficult to handle acids even in small amounts into solutions, its interaction with materials need to be taken into account in any hydrometallurgical application (Noor and Al-Moubaraki 2008). Methane sulfonic acid (MSA) is an organic acid formed by the atmospheric oxidation of dymethyldisulfide, this acid is readily biodegradable and decompose into sulphate, water, biomass and carbon dioxide. The lack of odour and the low possibility of generate harmful gases along with its low toxicity and corrosiveness make this acid a more “green” alternative for an acid leaching of chalcopyrite specially in In situ leaching applications (Hasan and Rohan 2010).
Even though the environmental benefits of this acid have been analysed for other applications (D. Gernon, Wu et al. 1999, Commarieu, Hoelderich et al. 2002, Hasan and Rohan 2010), the effectivity of MSA as lixiviant in hydrometallurgy processes is poorly studied, its high dissolving power, high conductivity strong behaviour compared to other organic acids, make it noticeable for the recovery of heavy metals such as lead and copper (Wu, Dreisinger et al. 2014, Feng, Wen et al. 2015). However its applicability in copper refractory minerals like chalcopyrite has not yet been tested. The present study aimed to understand the kinectics of the ferric clhoride with MSA systems in the dissolution of a natural sample with gangue inclusion to elucidate a possible application of this system for hydrometallurgical processes .
- Experimental
- Materials
All experiments were performed on chalcopyrite sample mineral (67.18% CuFeS2), the mineral was grinded by a ball mill, dry sieved into different grain size fractions ( ̴38 µm to ̴106 µm) and separated by riffle into representative fractions for initial analysis , the original sample was digested and analysed by elemental chemical distribution ICP–AES (Table 2). The Quantitative X-ray Diffraction (QXRD) analysis performed revealed that the sample consists of 67.18% chalcopyrite (CuFeS2), 24.42% quartz (SiO2) and 8.40% Sphalerite (ZnS) (Fig. 1).
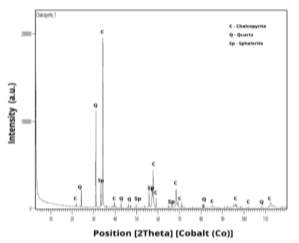
Fig. 1. X-ray difractogram of the initial sample containing Chalcopyrite, Quartz and Sphalerite
Table 2
Chemical composition of chalcopyrite sample -38µm
| Element | Si | Fe | Al | Ca | Cu | Zn | Ni | Pb | S |
| wt% | 12.1 | 19.4 | 0.423 | 1.39 | 21.9 | 4.99 | 0.833 | 0.054 | 24.4 |
Leaching stock solutions consisted in a combination of analytical grade ferric chloride Anhydrous from Chem Supply and 100% purity methanesulfonic acid (MSA) from BASF (Germany) to form ferric methanesulfonate dissolved in pure deionised water, the standard solution used had a total iron concentration of 3M and 0.05M free acid concentration this conditions were used as baseline for subsequent experiments with a selected parameter variation for each analysis. The initial pH was -2.44 and the Eh was 707 mV.
- Methods
The experiments were conducted in a closed rumbling water bath using 15 ml Nalgene bottles filled with 10 ml of solution and 1 gr of chalcopyrite. This bottles were placed inside 250 ml Nalgene heat resistant bottles filled with water at selected temperature to be tumbled at a constant rate of 15 inversions/min to maintain the heat transmission by fluid contact a constant stirring was selected to resemblance the possible scenario of an in situ leaching where the fluid is in continuous movement. The bottles and solutions were flushed with Nitrogen gas before closure to avoid oxygen inclusion in the system. Every point in the kinetic lines represents one individual experiment using the same conditions in order to avoid variations in the solid-liquid relation, the pH and Eh values were measured after every experiment to identify the acid and oxidant consumption, after leaching all the fluid was filtered and diluted for copper and iron final concentration analysis, the final content was determined using an Agilent 240/280 atomic absorption spectrometer (AAS) with a lamp current of 4 mA and 5 mA, and wavelengths of 222.6 nm – 217.9 nm and 386 nm – 392 nm for copper and iron respectively.
Final solids of each line were filtered, rinsed with matching background solution and dried using a vacuum filter and a standard oven, final solid weight was measured to keep control of the mass balance. The samples from the initial and final experiments were analysed to identify solid reaction-product phases with the PANalytical Empyrean X-ray diffractometer with Cu Kα1 radiation equipped with a PIXel detector, fixed 0.43° divergence slit at 40 kV and 45 mA and a step size increment of 0.0260 °2θ from 29 to 89 ° 2θ, the analysis of the acquired pattern was performed using High score plus 3.0d. Coarse size powder final solids (-106 + 75 µm) where processed by SEM-EDS with a JEOL-JSM-7001F field emission scanning electron microscope the set parameters were accelerating voltage of 20kV, 10mm working distance and emission current of 123.4mA, the resulting images were processed by Bruker ESPRIT 1.9. Final solids from selected lines were processed by ICP chemical analysis to identify leaching ratios.
To identify the influence of various parameters on leaching behaviour an standard line of experimental conditions was determined and used in most experiments (Table 3), just one of the parameters was change in each line, the remain conditions were sustained through the experiment .
Table 3
Base line operating conditions
| Chalcopyrite initial weight | 1 | gr |
| Solution volume | 10 | ml |
| Stirring speed | 15 | inv/mim |
| Particle size | – 45 +38 | µm |
| FeCl3 concentration | 3 | M |
| Acid (MSA) concentration | 0.5 | M |
| Solids loaded | 10 | % w/w |
| Temperature | 90 | ˚C |
- Results and discussion
- Initial acid concentration effect
The initial concentrations of MSA were change in a between 0.05 and 0.75 mol per litre keeping the designated standard conditions, Figure Xa indicate the comparative between the different MSA concentrations used for the chalcopyrite leaching, the effect of higher concentrations of acid is not significative however, the largest difference in leaching is located in between 0.1 and 0.5 mol per litre, in this point at 4 hrs there is a difference of 17.51% of copper recovery. No major differences were found in concentrations between maximum and minimum concentration both experiments reached the 100% recovery after 24 hr of leaching. From the final pH measurements was possible to see that the final values were still located under – 0.5 pH and the difference with the initial values did not exceed 1 point. This minor dependence of the leaching process on the initial acid concentration was seen in other copper bearing and lead minerals (Wu, Dreisinger et al. 2014, Feng, Wen et al. 2015) and represents an advantage in terms of acid usage in the reaction since it means that a similar leaching strength could be achieved with a minor concentration and that the gangue present in the initial sample did not interfere in a significant manner. The relation between MSA concentration and chalcopyrite dissolution delivered a value of power dependence of 0.18 which means that the dissolution is very independent of the initial acid concentration.
- Initial Iron (III) concentration effect
Reactions (1) to (3) represent the reaction between chalcopyrite and ferric chloride in an acid environment the initial formation of copper chloride complex by an intermediate oxidation results in the initial formation of a sulphur layer that covers the surface of the chalcopyrite, the entire oxidation involves a second stage of interaction between the sulphur formed and the remain ferric oxidant to release the final sulphate product into solution. For the final reaction 16 mol of ferric chloride are necessary for an entire oxidation of one mol of chalcopyrite, this means that for 1 gr of chalcopyrite 14.14 g of FeCl3 are required, nevertheless due to the final products of the leaching process in the present and other previous studies (Majima, Awakura et al. 1985, Havlík, Škrobian et al. 1995) it has been identified that the preferential reaction obeys to (1), in this case just 4 mol of ferric chloride are required for each mol of initial chalcopyrite sample, that means that a concentration of 2M is necessary as a started point for the leaching experiments under the standard conditions.
CuFeS
2
+4FeCl3
→CuCl2
+5FeCl2
+2S0 (1)
2S0+12FeCl3
+6H2
O →4SO42-+12Fe+2+16H++36Cl- (2)
CuFeS
2
+16FeCl3
+8H2
O →CuSO4
+FeSO4
+16FeCl2
+16HCl (3)
Concentrations between 1.5 and 3 mol per litre were selected to analyse the dependence of the copper recovery on the initial oxidant concentration, the results of four lines of experiments are shown in Fig. 2b. As expected the leaching rate increased with major concentrations of ferric chloride but in this case the dependence does not seems to be highly affected due to the high concentrations used, concentrations under 1 mol per litre are more sensitive to small changes as is possible to see in lower concentration plots (Hirato et al., 1986). All the tested concentrations reached the final dissolution after 24 hr of leaching but the final dissolution point was reached considerably more rapidly with the 3 mol per litre solution. Fig. 3 represents the relation between individual leaching rate constants and ferric chloride concentrations showing a linear pattern and a slope of 0.43 very similar number was reported by other authors going from 0.3 to 0.5 and 0.8 power dependence (Ammou-Chokroum, Sen et al. 1979, Dutrizac 1981, Majima, Awakura et al. 1985, Hirato, Kinoshita et al. 1986, Rath, Paramguru et al. 1988). The current work indicates that chalcopyrite leaching rate is nearly half order dependent on ferric chloride concertation, this value is considerably higher than the power dependence of acid initial concentration.
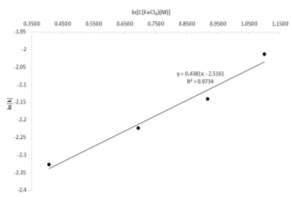
Fig. 3. Dependence of the dissolution rate of chalcopyrite leaching on ferric ion concentration.
- Particle size effect
Five different particle sizes were tested to identify the influence over the leaching process, I expected that the leaching rate have an inverse relation to grain size, due to the increase in the effective contact surface area between reactants, smaller the particle size greater is the probability that the collision of molecules will take place not only in the molecules on the surface. The sizes selected for the present study include a maximum size of -106 + 75 µm and a minimum also used for the standard lines of -38 µm, Fig. 2c shows that the response of the copper leaching is highly dependent on grain size small changes in size increase the difficulty of the leaching and the several extra hr of reaction, e.g. for the maximum size in the experiment 24 hr are necessary to reach 63% recovery and 120 hr to finalize the total dissolution, this strong influence differ from other studies under MSA environment (Wu, Dreisinger et al. 2014, Feng, Wen et al. 2015) but is well documented by other studies using ferric ion as an oxidant (Munoz, Miller et al. 1979, Rath, Paramguru et al. 1988, Al-Harahsheh and Kingman 2007).
- Temperature effect
The effect of temperature on experimental chalcopyrite dissolution is shown in Fig. 2d, the copper recovery was examined over a range of temperatures from 40 ᵒC to 90 ᵒC the influence of temperature appears highly marked between the minimum and maximum temperature tested, predictably the copper recovery will increase when the temperature increases, to the fact of this is an endothermic reaction and the energy input from heat transfer is a highly important contribution to the dissolution process. For 40 ᵒC experiments the maximum dissolution was not reached after 168 hr but the trend of the line suggest that the increasing behaviour will continue. The difference in time to reach the maximum dissolution between the 90 ᵒC and 70 ᵒC is 120 hr of experiment meaning that a small increment of temperature has a strong repercussion in reaction kinetics. This behaviour may the initial indicator of the reaction mechanism as chemical reaction control processes exhibit large differences between a short range of temperatures (Dreisinger and Abed 2002).
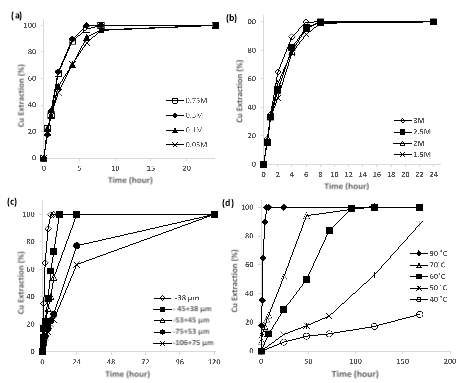
Fig. 2. Extraction of copper against reaction time showing the effect of operating parameters on chalcopyrite leaching: a. Initial acid concentration (mol/L), b. Initial iron concentration (mol/L), c. particle size (µm), d. Temperature(ᵒC)
- Kinetic analysis by shrinking core models
After the experiments testing different temperatures were done and made evident the big influence that this parameter has over copper leaching was necessary try to understand the dependence of the solid dissolution or transformation in function of the time, this relation is known as rate equation, Equation (4) represent the integrated rate equation were kc is the rate constant, t is the reaction time, x the fraction transformed and g(x) is the function that describe the variation of x with the time. In practice empirical functions are tested to find the reaction mechanism and determine the rate constant (Putnis 1992).
g(x)=kct (4)
Because the dissolution of chalcopyrite under acid conditions is a heterogeneous reaction, a shirking core model was selected, in this model the reaction front starts in the outer surface of the particle and is moving into the remaining bulk solid. The rate of the reaction is going to be controlled by one of successive steps during the reaction, this steps include the diffusion of the reactant to the particle surface through the surrounding solution, infiltration and diffusion of the reactant within pores to the mineral surface, surface chemical reaction with the solid and finally the transport of the products from the particle surface to the bulk solution (Yagi and Kunii 1955, Levenspiel 1999).
Empirical rate equations for heterogeneous reactions have been develop to support the reactions control steps, for the present study six of this equations were tried and evaluated based on its adjustment to the experimental data, the coefficient of determination, denoted R2 for each data set is shown in Table 4, the diffusion control through a product layer and fluid diffusion control kinetics did not fit properly to the temperature experimental data hence it was discarded, from the remained equations the highest regression coefficients were obtained by the chemical reaction control equation this values were consistent in all the parameters including acid and oxidant concentration (Fig. 4), this fact confirms the mechanism that was suggested by the important role of temperature in the experimental rates and is in agreement with other published kinetical analysis of ferric ion as oxidant (Dutrizac 1981, Palmer, Nebo et al. 1981, Majima, Awakura et al. 1985, Rath, Paramguru et al. 1988, Al-Harahsheh and Kingman 2007, Córdoba, Muñoz et al. 2008) .
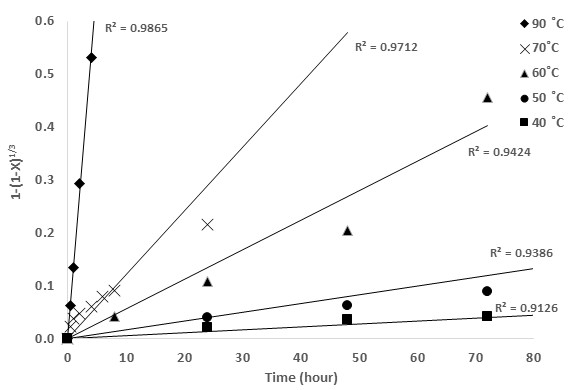
Fig. 4. Plot of fitting temperature experimental data by the chemical reaction control model 1-(1-X)1/3
Table 4
Comparative of values for correlation coefficient R2 of six kinetic experimental equations at test selected variables
| R2 | |||||||
| Film diffusion control |
Diffusion control through a product layer (cylindrical particles) |
Diffusion control through a product layer (spherical particles) |
Surface chemical reaction control (spherical small or large particles) |
Film diffusion control (spherical small particles) |
Film diffusion control (spherical large particle ) |
||
| Variable | X | X+(1-X)ln(1-X) | 1-3(1-X)2/3+2(1-X) | 1-(1-X)1/3 | 1-(1-X)2/3 | 1-(1-X)1/2 | |
| Fe3+ concentration (mol/L) | |||||||
| 3M | 0.7029 | 0.982 | 0.9617 | 0.9865 | 0.8696 | 0.9401 | |
| 2.5M | 0.8118 | 0.9677 | 0.969 | 0.9905 | 0.9422 | 0.9883 | |
| 2M | 0.7884 | 0.9861 | 0.9725 | 0.9973 | 0.9288 | 0.9777 | |
| 1.5M | 0.8343 | 0.9819 | 0.9624 | 0.9959 | 0.9477 | 0.9835 | |
| Particle size (µm) | |||||||
| 38 | 0.7029 | 0.982 | 0.9617 | 0.9865 | 0.8696 | 0.9401 | |
| 45 | 0.9753 | 0.898 | 0.8839 | 0.9891 | 0.9932 | 0.9901 | |
| 53 | 0.9523 | 0.9215 | 0.912 | 0.9792 | 0.9862 | 0.9746 | |
| 75 | 0.9161 | 0.9733 | 0.9711 | 0.9395 | 0.974 | 0.9341 | |
| 106 | 0.9682 | 0.9771 | 0.9755 | 0.9191 | 0.9755 | 0.9678 | |
| MSA concentration (mol/L) | |||||||
| 0.75M | 0.7029 | 0.982 | 0.9617 | 0.9865 | 0.8696 | 0.9401 | |
| 0.5M | 0.7029 | 0.981 | 0.9872 | 0.9945 | 0.8774 | 0.9537 | |
| 0.1M | 0.785 | 0.9808 | 0.9663 | 0.9828 | 0.9262 | 0.9568 | |
| 0.05M | 0.8206 | 0.9793 | 0.9642 | 0.9904 | 0.9308 | 0.9705 | |
| Temperature (ᵒC) | |||||||
| 90 | 0.7029 | 0.982 | 0.9617 | 0.9865 | 0.8696 | 0.9401 | |
| 70 | 0.9302 | 0.8996 | 0.8774 | 0.9712 | 0.9728 | 0.9772 | |
| 60 | 0.9914 | 0.7978 | 0.7741 | 0.9424 | 0.9761 | 0.9614 | |
| 50 | 0.9698 | 0.7323 | 0.7233 | 0.9386 | 0.956 | 0.9478 | |
| 40 | 0.8963 | 0.9743 | 0.9729 | 0.9126 | 0.9096 | 0.9086 | |
When a chemical reaction is the control step of the reaction the presence of the surface product layer does not affect the progress of the reaction and that the rate is proportional to the non-reacted core surface area and the reactant concentration in the bulk solution is equivalent to the concentration at the particle surface, Equation (5) describes the integrated rate equation for chemical reaction control processes for spherical shape particles (Levenspiel 1999), were X is the fraction of the reacted solid, τ is the time required to reach the complete conversion of the core (r=0), and kc is the kinetic rate constant.
tτ=kct=1-(1-X)1/3
(5)
X=1-rr0
(6)
kc=1τ=bCMkccρR
(7)
The temperature dependence of the rate constant in terms of the temperature and the energy needed to supply the reactants to form the activated complex know as activation energy (Ea) is represent by the Arrhenius law (8) to (10) (Levenspiel 1999). The rate constants (kc) for each temperature line were obtained from the slopes of the linear equations of the lines presented in Fig. 4, this values were used to obtain an Arrhenius plot. As shown in Equation (10) a plot of ln kc against 1/T provide a slope and intercept (1/T = 0) were the value of the apparent activation energy and the frequency factor (A) could be calculated (Fig. 5a).
kc=Ae-Ea/RT
(8)
lnkc=lnA-EaRT
(9)
Rearranging the equation for plotting purposes
lnkc= -EaR1T+lnA
(10)
The calculation of the other activation parameters were estimated using the transition state theory were the relation between temperature and reaction rate constant is given by Eyring equation (Kuhn 2000) the differential form of this equation relates the activation enthalpy term with the activation energy (11) to (13) .
kc=kbThe-∆G++/RT
(11)
dlnkdT=1T+∆H++RT2
(12)
Ea=∆H+++RT
(13)
The linearization of this equation (14) for practical effects allow us to determine the activation parameters (enthalpy and entropy) by a plot of ln (kc/T) against 1/T, where the slope gives the value of ∆H++ and the intercept on the abscissa the value of ΔS++ (Petek and Krajnc 2012) (Fig. 5b).
lnkT=
–
∆H++R1T+ lnkbh+∆S++R (14)
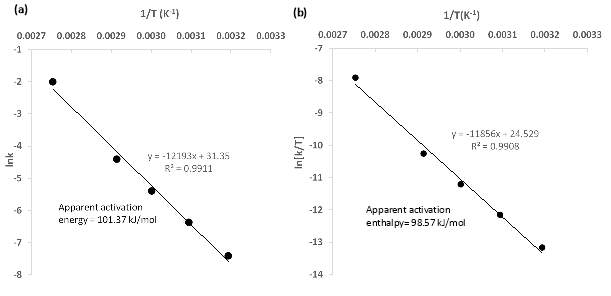
Fig. 5. Activation parameters determination a. Arrhenius plot for chalcopyrite dissolution using the data from experimental kinetic equation for chemical reaction control model for activation energy calculation, b. Eyring plot for chalcopyrite leaching for activation enthalpy calculation.
The apparent activation energy calculated by a shirking core model for the present study was found to be 101.37 kJ/mol. This value is consistent with the selected empirical equation to describe the mechanism of the reaction even though is above the range of 55 to 88 found by other authors with the same oxidant as previously described before (Table 1) however it is very close to the values of 131 kJ/mol and 93 kJ/mol found for ferric ion by Córdoba, Muñoz et al. (2008) and Rath, Paramguru et al. (1988). This values were attributed to the specific response of each sample to the chemical reaction, Dutrizac (1981) describes the content of gangue or non-copper minerals in the sample as a ‘complication factor’ with a high influence in the activation energy, due to the fact that in the present study the amount of non-copper minerals represents the 33% of the sample the higher value compared with the fairly pure chalcopyrite previous analysis seems valid. Leaving aside the particular nature of the sample the effort in carrying out the chemical reaction might reside in the crystal structure of chalcopyrite and its semiconductor nature, in the dissolution process major part of the energy implicated in the system is used in the consumption of an electron hole followed by an electron transfer step in the surface of the chalcopyrite which would explain the high energy value required (Hiskey 1993, Córdoba, Muñoz et al. 2008) likewise the S0 product of the reaction in chloride systems reported not to form a protective layer and to allow the continuous contact with the reactants since it has a porous morphology as demonstrated by previous studies (Palmer, Nebo et al. 1981, Hirato, Kinoshita et al. 1986, Rath, Paramguru et al. 1988) this supports the selection of chemical reaction as control of the process.
The frequency of the collisions a determinant factor of the reaction speed was found to be
4.1x1013h-1 indication of a slow process as evidenced in industrial practice. The value of activation enthalpy was determined to be 98.6 kJ/mol, the small difference between this value and the activation energy is in accordance with the temperature effect which in ambient temperature makes
Ea≅ ∆H++(13).
The estimate of the activation energy based on empirical mechanism equations has the disadvantage of been dependent on the selection of the rate function this can lead to a subjective selection when the fitting is similar for various equations, different values of rate constants have a large impact on the activation energy values, this and the impossibility of identify changes in energy values during the reaction suggest the necessity of an alternative methodology of verification. The independent method named ‘time to a given fraction’ is based on the relation between the reacted fraction x and the time t, turning time into a dependent variable (15) to (16).
dxdt=kc.f(x)
(15)
dt=kc-1.f-1x dx
(16)
Integrated between and initial and final fraction and with a constant function over the temperature range the final equation is a constant numerical value (17) to (19).
tx=kc-1 ∫x=0x=Xf-1xdx
(17)
tx∝A-1 e-EaRT
If
tx∝kc-1 (18)
lntx=const-lnA+EaR1T
(19)
Similarly to an Arrhenius plot the plot of ln tx against 1/T deliver a line with slope Ea/R from which it is possible to calculate the activation energy in an specific reacted fraction x, the calculation of the Ea in different fractions enables monitoring the possible change in the reaction mechanism (Putnis 1992, Qian, Li et al. 2017). In this study two fractions were analysed, the points situated at 20% and 50% represent the initial and middle life of the reaction were no points of inflexion were found (Fig. 6), the Ea values found were 101.21 kJ/mol and 94.95 kJ/mol respectively, the initial value is roughly the same as the average apparent activation energy obtained by the Arrhenius equation which confirms the choice of the chemical reaction as control step, the second value not far from the first one if the possible experimental errors involved are taken into account, indicating that the mechanism of reaction is maintained up to the half-life of the process and there are no substantial changes in the activation energy through the course of reaction. Table 5 summarizes the rate constants calculated for both methods for all the plots the linear regressions the experimental points fit properly and the correlation coefficients had values over 0.99.
Table 5
Kinetics rate constants for chalcopyrite dissolution in function of temperature for Arrhenius and ‘time to a given fraction’ methods
| T (ᵒC) | T (K) | 1/[T] (K-1) |
Rate constant k (h-1) |
ln k | ln (k/T) | Time to 50% Cu (h) |
Time to 20% Cu (h) | ln tx to 50% Cu (h) |
ln tx to 20% Cu (h) |
| 90 | 363.15 | 2.75E-03 | 1.34E-01 | -2.01 | -7.91 | 2 | 0.8 | 0.69 | -0.22 |
| 70 | 343.15 | 2.91E-03 | 1.21E-02 | -4.41 | -10.25 | 24 | 4 | 3.18 | 1.39 |
| 60 | 333.15 | 3.00E-03 | 4.50E-03 | -5.40 | -11.21 | 48 | 15 | 3.87 | 2.70 |
| 50 | 323.15 | 3.09E-03 | 1.70E-03 | -6.38 | -12.16 | 120 | 57 | 4.79 | 4.04 |
| 40 | 313.15 | 3.19E-03 | 6.00E-04 | -7.42 | -13.17 | 345 | 140 | 5.84 | 4.94 |
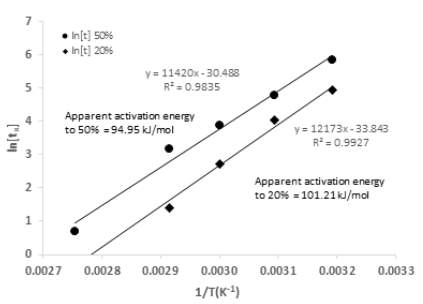
Fig. 6. Temperature data plotted as (time) to 50% and 20% of leaching of chalcopyrite (X=0.5 and X=0.2) against 1/T using the ‘time to a given fraction’ method for activation energy determination.
- Residue Characterization
Samples of the initial -106+75 µm chalcopyrite were examined by XRD and SEM, for X-ray analysis the samples were taken after 0.5 hr, 1 hr, 2 hr, 4 hr and 8 hr of leaching the resultant patterns are shown in Fig. 7, this comparative makes a sequence of appearance of the products of reaction were is possible to identify the new peaks of sulphur product in the range of 13 deg to 50 deg. The initial two main peak of chalcopyrite located at 34.24 deg is present in all the patterns nevertheless the intensity of the peaks decrease in around 74% between the initial and final point. Likewise is possible to identify the occurrence of sulphur main peak situated at 26.87 deg and its growing in the first 8 hrs, the disappearance of the initial chalcopyrite replaced by elemental sulphur may be the consequence of the morphology of the product observed by Dutrizac (1990) in small sixe particles where the formed sulphur wrap the chalcopyrite grain till the final dissolution.
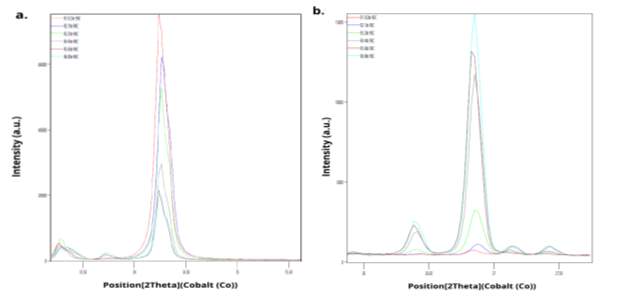
Fig. 7. X-raydifractograms showing the changes in peak intensity for six test times of reaction (0.5hr, 1hr, 2hr, 4hr, 6hr and 8hr) samples size -106+75 µma. Main chalcopyrite peak at 34.24 deg b. Main elemental sulphur peak at 26.87 deg.
The previous identification of sulphur as a reaction product was corroborated by scanning electron microscopy (SEM) on the samples, major part of the final solids were done in unpolished samples due to the fact that elemental sulphur tends to fall and re-group after polishing and due to the size of the analysed sample is difficult to appreciate any sulphur rim in the particle, Fig. 8a to Fig. 8c represent images of the unpolished surface of chalcopyrite grains after 8 hrs of leaching, is possible to see that the entire grain is covering in irregular interconnected globular grains of sulphur, the morphology of this layer appears to have intergranular porosity allowing the continuous flow between the bulk reactant and the chalcopyrite, as the leaching progresses, the open spaces of chalcopyrite decreases till be minimal as is evident in the micrographs, however as perceived in kinetical analysis the diffusion of the fluid through the product layer is not the rate control step of the reaction because the product layer is sufficiently porous and does not represent a barrier for continuous leaching (Majima, Awakura et al. 1985).
An interesting point found in the images (Fig. 8b) is the lack of surface layer covering quartz grains, this could be explained by a low wettability of silica with sulphur or that the formation of the of the sulphur forms by the attack of the FeCl3 directly in the surface of the chalcopyrite and notby the precipitation of species from the solution as presented by Dutrizac (1990), if that were the case the precipitation will cover all the other species present in the sample. The existence of cracks in the particles of chalcopyrite that don’t look like small pits developed by polishing is a possible evidence of reaction enhanced fractures as a result of the inclusion of sulphur during the reaction, in this manner the sulphur starts to fill the natural cracks and making them bigger by the dissolution of the chalcopyrite, This event would have repercussions on the accessibility of the fluid as it indicates that the solution is capable of entering the sample without major natural fractures.

Fig. 8. Scanning electron photomicrographs of -106+75 µm chalcopyrite after 8 hr leaching under base line conditions a. close up of a chalcopyrite particle, the particle is almost entirely wrapped by a sulphur layer, the sulphur layer form appears to be ‘cap head’ globular masses with spaces In between but interconnected. b. Chalcopyrite and quartz particles, the chalcopyrite is entirely cover by a sulphur layer and the quartz remained uncover. c. Covered particles of chalcopyrite where is possible to see the internal spaces in the sulphur that allows the flow between the sample and the bulk reactant. d. Epoxy mount sample of chalcopyrite after the sulphur layer is polished where internal cracks are visible.
- Concluding remarks
The kinetics of leaching for non-pure chalcopyrite sample with ferric chloride solution in an alternative methane sulfonic acid was studied by diverse parameters in a rumbling water bath with constant stirring. The findings of this study indicates that the concentration of methane sulfonic acid has a minimal influence on the leaching rate, minimal concentrations of acid are necessary to carry out the reaction, which represents an advantage in industrial applicability terms for this acid. The concentration of Fe+3 in the reaction neither does it seem to have an major influence in the kinetics, there is a dependence of around half order with ferric ions concentration but the changes on the leaching rate are not highly pronounced over the 1.5M concentration used in this study. Particle size and temperature resulted to have a major influence on kinetics, important changes in the reaction rate were found with small variations of time, by analysis of temperature data was possible to determine that the overall reaction is controlled by chemical reaction kinetics, fact that was corroborated by the calculation of the apparent activation energy by different methods, the average apparent activation energy was found to be 101.37 kJ/mol by Arrhenius equation, 94.95 kJ/mol and 101.21 kJ/mol for 20% and 50 % dissolution by the ‘time to a given fraction’ method. The selected reaction mechanism is an indication of the nature of the final product formed in the surface of the sample identified by X-ray diffraction as elemental sulphur and characterized by scanning electron microscopy as globular particles wrapping the entire particle of chalcopyrite with intergranular spaces that allowed the internal flow, furthermore the addition of MSA as lixiviant does not show evident influence in the texture or aspect of the final products. The kinetics of chalcopyrite with ferric chloride are consistent with previous studies, the change of acid does not have a greater impact on the reaction speed or the nature of the product, however for an ISR application the use of a biodegradable and recyclable acid, in conjunction with ferric ions and brine would constitute a reliable option for chalcopyrite extraction.
Acknowledgments
The funding for this study was provided by MRIWA and BASF and the Parker Centre for Integrated Hydrometallurgical Solutions. We would like to thank CSIRO mineral processing for the use of the installations
Nomenclature
t time
b stoichiometric coefficient
C bulk aqueous phase concentration
X fraction reacted
τ time to completely consume the particle
r radius of unreacted particle at time t
ρ molar density
M solid molar mass
kcc surface chemical rate constant
kc apparent reaction rate constant
r0 initial particle radius
Ea activation energy
A Preexponential factor
T recorded temperature
h Planck’s constant
kB Boltzmann’s constant
R ideal gas constant
ΔG++ Gibbs energy of activation
ΔH++ Enthalpy of activation
ΔS++ Entropy of activation
tx time to transform a given fraction
References
Al-Harahsheh, M. and S. Kingman (2007). “The influence of microwaves on the leaching of sphalerite in ferric chloride.” Chemical Engineering & Processing: Process Intensification 46(10): 1246-1251.
Al-Harahsheh, M., S. Kingman and S. Bradshaw (2006). “Scale up possibilities for microwave leaching of chalcopyrite in ferric sulphate.” International Journal of Mineral Processing 80(2): 198-204.
Al-Harahsheh, M., S. Kingman, N. Hankins, C. Somerfield, S. Bradshaw and W. Louw (2005). “The influence of microwaves on the leaching kinetics of chalcopyrite.” Minerals Engineering 18(13): 1259-1268.
Ammou-Chokroum, M., P. Sen and F. Fouques (1979). “Electro-Oxidation of Chalcopyrite in an Acid Chloride Medium. I.–Merit and Kinetics of the Reaction.” Mem. Sci. Rev. Metall., pp. 47, Apr. 1979: 47.
Commarieu, A., W. Hoelderich, J. A. Laffitte and M.-P. Dupont (2002). “Fries rearrangement in methane sulfonic acid, an environmental friendly acid.” Journal of Molecular Catalysis A: Chemical 182-183(Supplement C): 137-141.
Córdoba, E. M., J. A. Muñoz, M. L. Blázquez, F. González and A. Ballester (2008). “Leaching of chalcopyrite with ferric ion. Part I: General aspects.” Hydrometallurgy 93(3): 81-87.
Córdoba, E. M., J. A. Muñoz, M. L. Blázquez, F. González and A. Ballester (2008). “Leaching of chalcopyrite with ferric ion. Part II: Effect of redox potential.” Hydrometallurgy 93(3): 88-96.
D. Gernon, M., M. Wu, T. Buszta and P. Janney (1999). “Environmental benefits of methanesulfonic acid . Comparative properties and advantages.” Green Chemistry 1(3): 127-140.
Dreisinger, D. and N. Abed (2002). “A fundamental study of the reductive leaching of chalcopyrite using metallic iron part I: kinetic analysis.” Hydrometallurgy 66(1): 37-57.
Dutrizac, J. (1990). “Elemental sulfur formation during the ferric-chloride leaching of chalcopyrite.” Hydrometallurgy 23(2-3): 153-176.
Dutrizac, J. E. (1981). “The dissolution of chalcopyrite in ferric sulfate and ferric chloride media.” Metallurgical Transactions B 12(2): 371-378.
El Meray, M., K. Elamari, E. Jdid, P. Blazy, A. Bouhafid and I. Akalay (2000). “Acid and oxidizing leaching of chalcopyrite concentrate by ferric sulphate and fluorosilicic-nitric media.” Separation Science and Technology (USA) 35(13): 2143-2158.
Feng, Q., S. Wen, W. Zhao, C. LV and X. Bai (2015). “Leaching of Copper from Malachite with Methane-sulfonic Acid.” 22: 1037.
Hackl, R. P., D. B. Dreisinger, E. Peters and J. A. King (1995). “Passivation of chalcopyrite during oxidative leaching in sulfate media.” Hydrometallurgy 39(1): 25-48.
Hasan, M. and J. F. Rohan (2010). “Cu electrodeposition from methanesulfonate electrolytes for ULSI and MEMS applications.” Journal of the Electrochemical Society 157(5): D278-D282.
Havlík, T., M. Škrobian, P. Baláž and R. Kammel (1995). “Leaching of chalcopyrite concentrate with ferric chloride.” International Journal of Mineral Processing 43(1-2): 61-72.
Hirato, T., M. Kinoshita, Y. Awakura and H. Majima (1986). “The leaching of chalcopyrite with ferric chloride.” Metallurgical Transactions B 17(1): 19-28.
Hirato, T., H. Majima and Y. Awakura (1987). “The leaching of chalcopyrite with ferric sulfate.” Metallurgical Transactions B 18(3): 489-496.
Hiroyoshi, N., H. Miki, T. Hirajima and M. Tsunekawa (2001). “Enhancement of chalcopyrite leaching by ferrous ions in acidic ferric sulfate solutions.” Hydrometallurgy 60(3): 185-197.
Hiskey, J. (1993). Chalcopyrite Semiconductor Electrochemistry and Dissolution. The Paul E.Queneau International Symposium. Extractive Metallurgy of Copper, Nickel and Cobalt. 1: 949-969.
Kaplun, K., J. Li, N. Kawashima and A. R. Gerson (2011). “Cu and Fe chalcopyrite leach activation energies and the effect of added Fe3+.” Geochimica et Cosmochimica Acta 75(20): 5865-5878.
Kuhn, H. (2000). Principles of physical chemistry : understanding molecules, molecular assemblies, supramolecular machines. Chichester, U.K., Chichester, U.K. : John Wiley.
Levenspiel, O. (1999). Chemical reaction engineering / Octave Levenspiel. New York, New York : Wiley.
Lu, Z. Y., M. I. Jeffrey and F. Lawson (2000). “The effect of chloride ions on the dissolution of chalcopyrite in acidic solutions.” Hydrometallurgy 56(2): 189-202.
Lundstrom, M., J. Aromaa, O. Forsen and M. Barker (2008). “Reaction product layer on chalcopyrite in cupric chloride leaching.” Canadian Metallurgical Quarterly 47(3): 245-252.
Majima, H., Y. Awakura, T. Hirato and T. Tanaka (1985). “The Leaching of Chalcopyrite in Ferric Chloride and Ferric Sulfate Solutions.” Canadian Metallurgical Quarterly 24(4): 283-291.
McDonald, R. G. and D. M. Muir (2007). “Pressure oxidation leaching of chalcopyrite. Part I. Comparison of high and low temperature reaction kinetics and products.” Hydrometallurgy 86(3): 191-205.
Munoz, P. B., J. D. Miller and M. E. Wadsworth (1979). “Reaction mechanism for the acid ferric sulfate leaching of chalcopyrite.” Metallurgical Transactions B 10(2): 149-158.
Nazari, G. and E. Asselin (2009). “Morphology of chalcopyrite leaching in acidic ferric sulfate media.” Hydrometallurgy 96(3): 183-188.
Noor, E. A. and A. H. Al-Moubaraki (2008). “Corrosion behavior of mild steel in hydrochloric acid solutions.” International Journal of Electrochemical Science 3(7): 806-818.
Northey, S., N. Haque and G. Mudd (2013). “Using sustainability reporting to assess the environmental footprint of copper mining.” Journal of Cleaner Production 40(Supplement C): 118-128.
Olvera, O. G., M. Rebolledo and E. Asselin (2015). “Atmospheric ferric sulfate leaching of chalcopyrite: Thermodynamics, kinetics and electrochemistry.” Hydrometallurgy.
Orth, R. J. and K. C. Liddell (1990). “Rate law and mechanism for the oxidation of copper(I) by iron(III) in hydrochloric acid solutions.” Industrial & Engineering Chemistry Research 29(7): 1178-1183.
Palmer, B. R., C. O. Nebo, M. F. Rau and M. C. Fuerstenau (1981). “Rate phenomena involved in the dissolution of chalcopyrite in chloride bearing lixiviants.” Metallurgical Transactions B 12(3): 595-601.
Petek, A. and M. Krajnc (2012). “The enthalpy and entropy of activation for ethyl acetate saponification.” International Journal of Chemical Kinetics 44(10): 692-698.
Putnis, A. (1992). Introduction to mineral sciences, Cambridge University press.
Qian, G., Y. Li, J. Li and A. R. Gerson (2017). “Consideration of enthalpic and entropic energy contributions to the relative rates of chalcopyrite dissolution in the presence of aqueous cationic impurities.” International Journal of Mineral Processing 159: 42-50.
Rath, P. C., R. K. Paramguru and P. K. Jena (1988). “Kinetics of dissolution of sulphide minerals in ferric chloride solution, 1: Dissolution of galena, sphalerite and chalcopyrite.” 97: 150-158.
Saxena, N. N. and N. R. Mandre (1992). “Mixed control kinetics of copper dissolution for copper ore using ferric chloride.” Hydrometallurgy 28(1): 111-117.
Seredkin, M., A. Zabolotsky and G. Jeffress (2016). “In situ recovery, an alternative to conventional methods of mining: Exploration, resource estimation, environmental issues, project evaluation and economics.” Ore Geology Reviews 79: 500-514.
Shiers, D. W., D. M. Collinson, N. J. Kelly and H. R. Watling (2016). “Copper extraction from chalcopyrite: Comparison of three non-sulfate oxidants, hypochlorous acid, sodium chlorate and potassium nitrate, with ferric sulfate.” Minerals Engineering 85: 55-65.
Wang, S. (2005). “Copper leaching from chalcopyrite concentrates.” JOM 57(7): 48-51.
Wu, Z., D. B. Dreisinger, H. Urch and S. Fassbender (2014). “The kinetics of leaching galena concentrates with ferric methanesulfonate solution.” Hydrometallurgy 142: 121-130.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Chemistry"
Chemistry is a science involving the study of the elements and matter at the atomic and molecular level including their composition, structure, properties, behaviour, and how they react or combine.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: