Digestive Enzyme Therapy Used in Metabolic Pathologies
Info: 8505 words (34 pages) Dissertation
Published: 16th Feb 2022
Tagged: Biomedical Science
Abstract
Enzymes have high catalytic efficiency and a great affinity. They are needed for all the chemical inter conversions which are required to support life and hasten the metabolic processes taking place in the body. It is these properties which make them different when compared to other types of drugs (Schibli et al. 2002); Enzymes are used in different therapeutic purposes. Digestive and metabolic enzymes are used with other therapies in the treatment of illnesses such as skin ulcers, leukemia, ulcers, Pompe’s disease, cardiovascular diseases, celiac disease, Parkinson’s disease, Fabry’s diseases, inflammation, pancreatic disorders and digestive disorders (Gonzalez et al. 1995; Schibli et al. 2002). Enzymes are also used in the diagnosis, monitoring and investigation of life threatening diseases. Medically important enzymes which have been produced by microorganisms are more consistent and more effective. The present paper will look into specific digestive enzyme therapy used in metabolic pathologies particularly in neonates and infants, human rhDNase therapy for Cystic Fibrosis and its associated RTIs and Asparaginase.
Keywords: Enzymes, Recombinant human deoxyribonuclease 1, Cystic Fibrosis
List of Abbreviations
EcAII – Escherichia coli
SER – Smooth Endoplasmic Reticulum
CF- Cystic Fibrosis
CFTR – Cystic Fibrosis Transmembrane Conductance Regulator
RhDNase – Recombinant human deoxyribonuclease 1
ErA – Erwinia chrysanthemi
GALT – Galactose-1-phosphate uridyl transferase
Table of contents
Click to expand Contents
Introduction
Specific digestive enzyme therapy used in metabolic pathologies particularly in neonates and infants.
Human rhDNase therapy for Cystic Fibrosis and its associated RTIs
Asparaginase
Conclusion
References
List of Figures
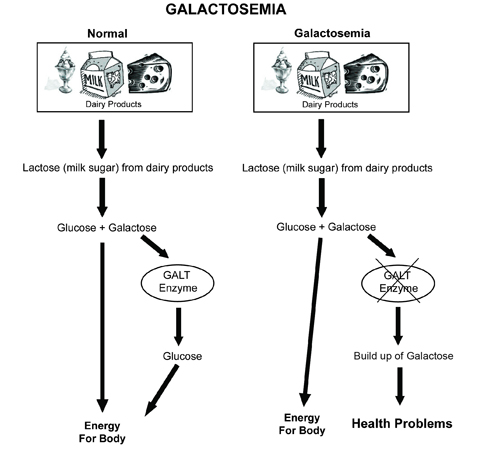
Figure 1: Galactosemia due to lack of the GALT enzyme
Figure 2. Schematic illustration of the types of bonds occurring in mucous gel, illustrating potential targets for mucoactive agents
Figure 3. Schematic illustration of the types of bonds occurring in mucous gel, illustrating potential targets for mucoactive agents
Figure 4: DNase which enzymatically cleaves extra cellular DNA into shorter molecules
Figure 5: The addition of DNase to sputum
Figure 6: Image showing DNase depolymerizing sputum DNA filaments
List of Tables
Table 1: Enzymes and their therapeutic use
Table 2: Functions of Enzymes in Human Milk
Table 3: Table showing some of the enzymes present in infants
Table 4: Incidence of rhDNase-Related and Cystic Fibrosis–Related Adverse Events
Table 5: Microbial therapeutic enzymes and their application
Introduction
Therapeutic enzymes are enzymes which are used medically or adjunct together with other enzymes for the treatment of diseases (Hwang et al.2013). Some of the therapeutic uses of enzymes include:
Table 1: Enzymes and their therapeutic use
| Enzymes | Therapeutic Use | Basis |
| Prolactazyme | Used for lactose intolerance | This is a proenzyme which is responsible for the production of lactase in the stomach |
| Algucerase | Gaucher’s Disease type 1 | It is formed when penicillin is converted to penicillioate |
| Asparaginase | Acute Childhood Leukemia | It reduces the level of serum asparagine and prevents the increase of asparagine dependent tumor cells |
| DNase | Cystic Fibrosis (CF) | It hydrolyses extracellular DNA which causes Cystic Fibrosis |
| Trypsin | Inflammation | Helps in the conversion of urate to allantoin |
| Enzyme inhibitors | To increase drug efficacy | It fights against resistant bacterisa |
| Ribonuclease | Antiviral Therapy | It hydrolyses RNA |
Specific digestive enzyme therapy used in metabolic pathologies particularly in neonates and infants
Enzymes are proteins which have unique abilities. They are found in living cells and are crucial for chemical reactions taking place in the body. Human milk has many enzymes which are of great benefit to the infant and they all have different functions (Gonzalez et al. 1995)
Table 1: Functions of Enzymes in Human Milk (Gonzalez et al. 1995; Colboum et al. 2007; Love et al. 1979)
| Function | Enzymes | Process |
| Biosynthesis of the different components of milk that are present in the mammary gland | Phosphoglucomutase | Main role is to synthesize lactose |
| Lactose synthetase | Lactose synthesis | |
| Fatty acid synthetase | Synthesis of medium-chain fatty acids | |
| Lipoprotein lipase | Regulate the circulation of triglyceride fatty acids | |
| Digestive function of the infant | Amylase | Assist in the hydrolysis of polysaccharides |
| Lipase | Help in the hydrolysis of triglycerides | |
| Proteases | Helps in the proteolysis process | |
| Transportation in the infant | Xanthine oxidase | It transports ion and molybdenum around the body |
| Glutathione peroxidase | Transports selenium ions | |
| Alkaline phosphatase | Facilitate transportation of magnesium and zinc ions | |
| Preservation of the different components of milk | Antiproteases | Protects immunoglobulin and other bioactive proteins |
| Sulfhydryl oxidase | It helps in the maintenance of the structure of proteins which have S-S bonds | |
| Anti-infective agents | Lysozyme | Bactericidal |
| Peroxidase | Bactericidal | |
| Lipases | Help in eliminating fatty acids which have antiviral, antibacterial and antiprotozoan actions | |
| Protect the body against enterocolitis | PAF-AH | Main function is to hydrolyze platelets responsible for necrotizing activities |
Antiprotease helps in the protection of the mammary gland from proteolysis. Proteolysis is as a result of lysosomal proteases. They also ensure that proteolytic breakdown of milk proteins does not reach so that they can reach the infant while they are still intact (Ito et al. 1984; Wong et al. 2012). The enzymes in the human milk have antitryptic and antichymotryptic abilities and, as such, ensure that there is no absorption of endogenous and bacterial proteases in infants. This helps in the protection of other extra intestinal organs, for example, the liver.
The high activity of the Antiproteases taking place in the colostrums happens just as nonimmunoglobulin protein is being transferred from the intestine to the systemic circulation of the infant. The digestive enzymes that are present in milk, digestive lipase and amylase, play the role for the pancreatic which is yet to be fully developed in the infant. Amylase and lipase are stable and can stay for many years (Blake et al.2007). The antiprotease activity of milk could be the reason for the high stability of enzymes present in milk.
Amylase is crucial to the infant especially after it starts using starch supplements. It is also important when the formula having oligosaccharides which have been hydrolyzed by amylase is given to the infants who are still breastfeeding. Compared to the amylase activity in adults, which of infants is below 0.5%. Even after breastfeeding for a period of four to six months, the infant lacks endogenously produced amylase. This is produced from the salivary glands (Berseth et al.1996; Gonzalez et al. 1995). The adequate level needed for pancreas enzymes in an infant take about two years after the birth of the infant. Milk amylase is also important for infants who are suffering from pancreatic insufficiency that is as a result of cystic fibrosis and malnutrition. Milk amylase has been studied extensively in the last ten years due to its ability to compensate for the low pancreatic lipase in infants because of the bile salt-dependent lipase found in the enzyme.
Another enzyme that is nutritionally important for the fetus and the neonate is Arginine. It is important since it detoxifies ammonia and helps in the synthesis of molecules which are of great significance to the fetus. Some of these molecules include nitric acid, creatine and polyamines (Marini et al. 1987). One of the major causes of nutritional problems in preterm infants is the lack of Arginine (hypoargininemia) in their developing bodies. The lack of hypoargininemia in the infant’s body leads to hyperammonemia dysfunction. Other related nutritional problems because of lack of this enzyme include pulmonary, intestinal, cardiovascular and neurological dysfunctions. In addition to that, it may result in high rates of infant morbidity and mortality which is due to premature births (Karlson et al. 1993). Intestinal citruline and arginine synthesis (which is one of the main sources of arginine) is not adequately present in preterm neonates because of the limited expression of genes required for major enzymes such as lyase, pyrroline-5-carboxylate synthase and argininosuccinate synthase among others. Consequently, when these enzymes lack in the body, it leads to hypoargininemia. It is important to note that premature births on women usually happen prior to the normal perineal surge of cortisol (Cortisol is an inducer and its representative of major arginine-synthetic enzymes) (Nussbaum, 1985; Gonzalez et al. 1995; Colboum et al. 2007; Love et al. 1979). It is therefore important since it helps in the maturation of intestinal arginine synthesis present in neonates. There are other advantages for the administration of the cortisol treatment. Some of these advantages are; it helps in enhancing and promoting enteral feeding to preterm infants and this is important for the synthesis of arginine, citruline and polyamines taking place in the intestines. Secondly, it enhances the growth of the intestines, the integrity and motility of the intestines. Lastly, it helps organs to mature faster while restoring the full enteral system of feeding.
Table 2: Table showing some of the enzymes present in infants (Barton et al. 1991)
| Term | Preterm infants | ||
| Mother’s milk | Enzymes present | Proteases (Arionic, typsin, anionic, eslastase, plasminogen, plasmin and plasminogen activator, cathepsin D-like protease)
Antiproteases (antitrypsin and antichymotrypsin) |
Proteases undetermined
Antiproteases (antitrypsin and antichymotrypsin) |
| Mouth | Enzymes present | Undetermined | Undetermined |
| Stomach | Enzymes present | Pepsin | Pepsin |
| Protein Degradation | Little to none | Little to none, 15% at 5-6 weeks postpartum (average 29 weeks gestational age) | |
| Small intestine | Enzymes present | Trypsin, chymotrypsin, elastase, enterokinase and carboxypeptidase B | |
| Protein Degradation | Undetermined | Undetermined | |
| Large intestine | Enzymes present | Protein-fermenting bacteria (including bifidobacterium longum subsp infantis) | Protein-fermenting bacteria (including bifidobacterium longum subsp infantis ) |
| Protein Degradation | 46-63% no longer intact in feces, 37-45% remains intact | Undetermined |
Galactose-1-phosphate uridyl transferase (GALT) is also an important enzyme in infants. The main function of this enzyme is to convert galactose to glucose. It becomes impossible to convert galactose to glucose when the GALT enzyme is not working properly in the body or it is lacking and this results in the building up of galactose in the blood (Barker, 2002). So that the body can use the various sugars and carbohydrates taken by the infant, the GALT enzyme breaks these sugars down to glucose for the body to use. Lactose is the sugar that is present in milk and it comprises of a single molecule of galactose and another one of glucose. When digestion is taking place, lactose is broken down into glucose and galactose. Lack of GALT enzyme in the infants body results in galactosemia. Galactosemia is a situation where the blood contains galactose. Galactose affects several organs in the body including the kidneys, eyes, brain and the liver. Infants suffering from galactosemia usually experience diarrhea and when they drink milk, they end up vomiting (Du et al. 2005; Slattery et al. 2001). Other effects of galactosemia are irritability, redundant and stunted weight and growth, lethargy and poor sucking and poor feeding practices. If the problem is not dealt with, it results in early cataracts in the child, jaundice (whitening of the eyes and yellow patches appearing on the skin), a condition known as hypoglycemia, an enlarged and ineffective liver, bleeding and blood infections which can result in death and shock.
Figure 1: Galactosemia due to lack of the GALT enzyme
Neonates and infants usually have immature organ systems, more so the liver, the lungs and the kidneys. These are the most commonly used routes for drug excretion in infants. There is a large surface to mass ratio and the high rates of metabolism in infants and neonates have several implications (Gonzalez et al. 1995; Funakoshi et al. 1977). There are enzymes in the liver which facilitate the metabolism processes for drugs and these are depressed in the newborn. In an adult, oxidation and conjugation leads to water soluble metabolites which are be excreted into the lungs easily. However, in the fetus, all these processes are suppressed and it is beneficial since lipid soluble metabolites can be transferred easily to the placenta where they are handled by the maternal system. The location of these hepatic microsomal enzymes is the liver smooth endoplasmic reticulum (SER). The liver of a neonatal has very little smooth endoplasmic reticulum and therefore, many activities of microsomal enzymes are reduced or rendered undetectable (Murai et al. 1978).
Human rhDNase therapy for Cystic Fibrosis and its associated RTIs
Cystic Fibrosis (CF) is one of the most common and well-known genetic disorders. It is one of the most dangerous autosomal recessive disorders and it affects several organs in the body. CF affects the sweat glands, the pancreas and the lungs among other body organs (Kerem et al. 1989; Tsui, 1992; Randell et al. 2006). The main cause of CF is a defective gene which results in the accumulation of a thick mucus layer. This defective gene is referred to as the CFTR gene, an encoding for a protein known as Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). CFTR is a transmembrane channel which is found in the epithelial cells and its main function is to control the movement of bicarbonate and chloride ions in and out of the cells (Carroll et al. 2002). When these genes are not working, the CFTR is not going to function and, as such, the person will start showing symptoms and signs of CF. When there are mutations in the CFTR gene, this can lead to several defects in some regions such as the regulation, the stability, synthesis, functioning and regulation of the chloride ion channel. The CFTR chloride channel is present at the apical surface of epithelial cells, epithelial cells, intestinal epithelium, vas deferens and the biliary ducts (Riordan et al. 1989). The disease is responsible for abnormal ion transportation through the epithelial membranes and, consequently this leads to thick tenacious secretions (Hwang et al. 2013). This mucus layer blocks the pancreas and the air paths in the body. Mucus is a heterogeneous and viscoelastic gel which consists of glycoprotein forming an entangled three-dimensional network on bonds (Derichs, 2013). This 3D structure is what makes the gel to be viscoelastic. In several respiratory infections, high concentrations of DNA released from degenerating polymorphonuclear leucocytes can be demonstrated in mucus and are associated with higher mucus viscosity.
Figure 3. Schematic illustration of the types of bonds occurring in mucous gel, illustrating potential targets for mucoactive agents
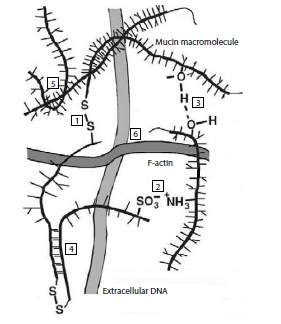
| Type of bond | Mucoactive agent |
| 1. Covalent bonds: glycoprotein subunits are linked primarily by intramolecular disulfide bonds | N-acetylcysteine Dithiothreitol |
| 2. Ionic bonds: mucin macromolecules have both positive and negative fixed charges capable of interacting | Hypertonic saline Dextran sulfate Heparin |
| 3. Hydrogen bonds link neighboring oligosaccharide side chains | Mannitol Dextran |
| 4. Van der Waals’ forces: bonds are due to van der Waals attractive forces between complementary saccharide moieties on neighboring chains | |
| 5. Intermingling: physical entanglements between mucin | High frequency oscillation |
| 6. Extracellular DNA and F-actin: parallel network formation due to infection | rhDNase Gelsolin |
Figure 4: DNase which enzymatically cleaves extra cellular DNA into shorter molecules 1. Bacteria grow in the mucous, causing inflammation, 2. Neutrophils are sent in response to inflammation, 3. As neutrophils die, elastase is released which damages lung cells, 4. DNA is also released, which thickens mucous, 5. Pulmozyme reduces the thickness of the mucous by breaking down DNA.

Recombinant human DNase (rhDNase)
Purulent sputum from patients with CF and several other respiratory diseases contains high concentrations of DNA which are produced by degenerating polymorphonuclear leucocytes. Higher DNA content in CF mucus is what leads to the higher mucus viscosity and mucus elastic modulus (Patel et al. 2008; Durward et al. 2000). The addition of exogenous DNA to sputum increases both viscosity and elasticity.
Purulent sputum has both DNA and a large volume of broad-spectrum protease; these two are products of neutrophils. DNA ensures that the protease does not destroy the mucin (Slattery et al. 2000). The vulnerability of mucin is increased upon the removal of DNA and as such, it becomes prone to protease attack. It is then hydrolyzed by protease, resulting to mucolysis. RhDNase greatly reduces viscosity of purulent CF-sputum in a concentration-dependent manner (Aitken et al. 1992; Barton et al. 1991). This reduction is associated with shortening of DNA-fragments in sputum.47 rhDNase also improves surface properties of CF sputum, as demonstrated by a decrease in the contact angle. In CF, rhDNase is also able to increase the free water content and alter the phospholipids profile of mucus, with a related improvement in mucus transportability.
There are over 1500 mutations of the CFTR gene that are currently known. Of all these mutations, the most common is a three base-pair deletion which leads to deletion of the phenylalanine residue at amino acid position 508 (this is known as the delta F508) (Bartelink et al. 2006; Blake at al. 2007). All these mutations can be divided into five main groups. Their categorization is based on their predicted and possible functional repercussions. Class I-111 is considered as severe mutations and they do not produce any functioning CFTR protection. The second category is class IV and V mutations which lead to mild pancreatic diseases. There are a few functioning CFTR proteins but they are in very small quantities. The first category of Class 1-111 is associated with severe clinical phenotype but genotype-phenotype relationship is variable with crucial heterogeneity in severity of lung disease (Ramjeesingh et al. 2003). The most lethal manifestation of cystic fibrosis is chronic progressive lung cancer. Though there are antibiotic therapies and chest physiotherapies which try to deal with the problem, it is one of the causes of disability and death. In every 2500 births, the occurrence of CF is one (Riordan, 2008).
It is however important to note that this ration accounts for the defective genes which are present in homozygous people. Most individuals are born with a defective CFTR gene and, they do not know (Hwang et al. 2013). A lot of research has been done on CFTR and CF and, this has resulted in several treatment procedures and possible cures. Airway secretions play a significant role in the respiratory dysfunction in cystic fibrosis. The thick tenacious secretions are viscous and it is not easy to expectorate them. Consequently, they block the airways and thus reduce the volume of air in the lungs while cutting down the expiratory flow rates (Randell, 2006). CFTR genes have very high concentrations of DNA (more than fifteen milligrams per milliliter) and this makes them very viscous and tenacious. These DNA are derived from disintegrated inflammatory cells in the body. Naturally, DNA has a tendency of forming viscous, thick, purulent secretions in cystic fibrosis which have same features as those present in conc. solutions of DNA (Kerem et al. 1989; Tsui, 1992). Even through mucus obstruction is believed to the main cause of CF lung disease, the chronic infection of the respiratory tract is a more destructive and dangerous process. The excess mucus becomes a breeding ground for the growth of pathogenic organisms such as Pseudomonas aeruginosa, Staphylococcus aureus and Haemophilus influenza and, once established, these organisms become impossible to get rid of. To prevent the damaging of the lungs, it is imperative to eliminate as much sputum as possible from the lungs every day.
Recombinant human deoxyribonuclease 1 (rhDNase) provides a solution through cleaving and removing the DNA present in purulent lung sections. RhDNase is an enzyme which has the ability of breaking down DNA strands found in air path secretions. RhDNase also has the ability of hydrolyzing the DNA that is in the mucus of a cystic fibrosis patient (Shak et al. 1990; Gadsby et al. 2006; Carroll et al. 2008). It can therefore be used with patients who have cystic fibrosis in eliminating all the secretions present in the lungs. In vitro, rhDNase cleaves DNA that is in the sputum in cystic fibrosis patients. It helps in reducing and lowering the sputum viscosity (Andersen, 2000). RhDNase acts in vivo to cleave high-molecular-weight DNA in purulent lung secretions and as such, improve the functioning of the lungs.
Figure 5: The addition of DNase to sputum has the effect of converting thick gel like sputum (A) into liquid (B)
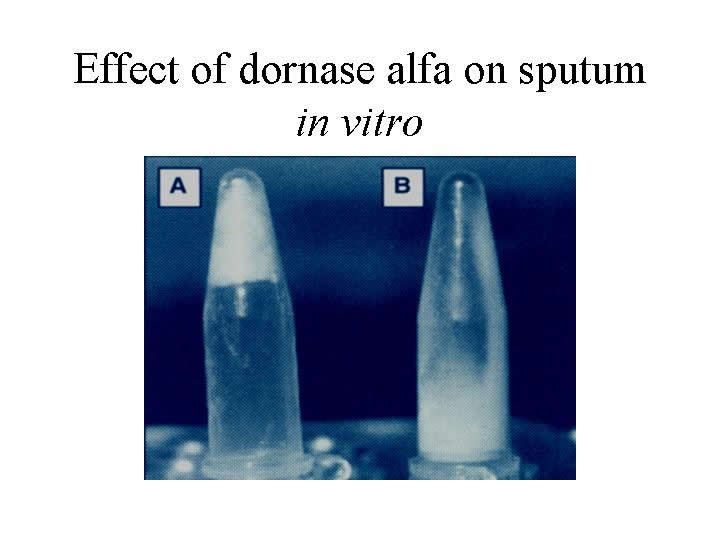
Treatment through the use of aerosolized rhDNase is an effective and efficient way of improving and enhancing the lung functions of patients suffering from cystic fibrosis (Durward et al. 2000). The main advantages of aerosolized rhDNase are that they are administered easily, are well tolerated by the patient’s body and improve the lung functioning within a short period of time (Greally, 1995). However, rhDNase is likely to be effective in patients who have purulent sputum. It enhances the elimination of small quantities of purulent mucus present in sites which are vital for the functioning of lungs and in areas where normal cough and mucociliary mechanisms are not able to get rid of thick and purulent mucus which has been trapped. It reduces the viscosity in the lungs while ensuring that secretion is being removed (Puterman et al. 1997; Patel et al.2000).
Biological rationale for rhDNase therapy
As explained above, the rationale behind the use of the rhDNase therapy relates to the fact that purulent pulmonary secretions have high concentrations of extracellular DNA. The DNA is produced due to the disintegration of neutrophils. According to experiments conducted by Potter et al. (1969), the results showed the presence of DNA in purulent in great amounts (4-15 mg/mL) (Funakoshi et al. 1997). DNA is a viscous and heavy polyanion; it is one of the contributing factors to the great viscosity of the secretions of the lung. In another experiment by Chernick et al (1961), lung secretions which had been incubated in vitro with partially bovine pancreatic DNase indicated a drop in their levels of viscosities. Bovine pancreatic DNase 1 (dornase) started being used officially in 1958. Clinical studies with patients who had pneumonia showed that the use of dornase was safe and it could reduce the viscosity of lung secretions (Wong et al. 2005).
Figure 6: Image showing DNase depolymerizing sputum DNA filaments
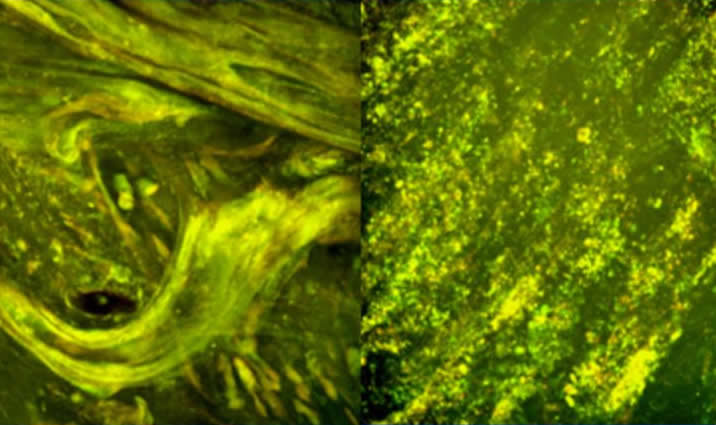
Despite it being approved for use, severe adverse respiratory reactions were reported at times, mostly due to the allergic reactions to a foreign protein. The gene for human DNase underwent a cloning process from a pancreatic cDNA library. The recombinant human DNase (rhDNase) was then synthesized. Human DNase and rhDNase are identical; it is therefore safe to use the latter without risking the consequences that come with the use animal proteins (Shah et al. 1996; Greally, 1995; Riordan, 2008;Murai et al, 1978) . Catalytic amounts of rhDNase have the ability of reducing the viscosity of purulent CF sputum. RhDNase converts the viscous purulent sputum to a flowing light liquid in a few minutes. For patients suffering from CF, treatment with rhDNase for a period of one and a half years reduces the DNA load present in the BAL fluid. This helps in clearing the lower airway secretions.
Even though rhDNase was developed for people who were suffering from cystic fibrosis, it can also be used as a rational therapy for lung diseases and tract infections which entail the plugging of mucus and impaired mucociliary clearance, where there is presence of netrophilic airway inflammation or increase in the amount of mucus in the DNA (Greally, 1995). Netrophilic inflammation is seen in adults with stable and acute asthma in asthmatic children in infants with respiratory syncytial virus (RSV) bronchiolitis. However, during a rhDNase therapy, some patients experience respiratory tract infections (RTI). These are characterized by several symptoms such as increased coughs, breathlessness, and the production of sputum and decreased functioning of the lung (Patel et al. 2000).
Karsten et al (1998) conducted a multinational, open-label study of 974 cystic fibrosis patients who had mild lung diseases. The study aimed at assessing the safety and efficacy of aerosolized rhDNase, 2.5 mg, once daily over a period of at least 12 weeks. The results were as follows:
Table 3—Incidence of rhDNase-Related and Cystic Fibrosis–Related Adverse Events
During the First 12 Weeks of Treatment
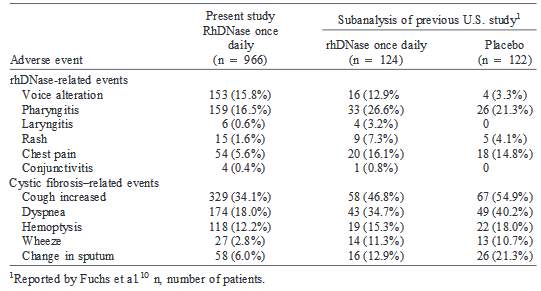
Even with antibiotic therapy, chronic RTIs are still a main cause of morbidity in patients with cystic fibrosis. The bacteria responsible are mostly P. aeruginosa, S. aureus, and Haemophilus influenzae.
Asparaginase
Asparaginase is an enzyme which is used as a medication and food processing. It is also referred to as crisantaspase. Asparaginase is used in the treatment of acute lymphoblastic leukemia (ALL), non-Hodgkin’s lymphoma and acute myeloid leukemia (AML). It is administered through an injection under the skin or through a vein (Amena et al. 2010; Boyse et al. 1967). Asparaginase breaks down amino acids which are known as asparagine. If the amino acid is not present, it becomes impossible for cancer cells to make DNA. Asparaginase (L-asparagine amidohydrolase, EC 3.5.1.1) is an enzyme whose major function is to catalyze and enhance the conversion and transformation of L-asparagine to L-aspartic acid and ammonia (Basha et al. 2009).
The asparaginase reaction:
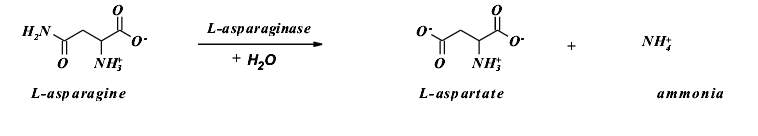
The substrate and the product of this enzymatic reaction play crucial roles in the metabolic processes taking place in living organisms, in animals, human beings and bacteria. In plants, L-asparagine is the most abundant metabolite and it stores and transports nitrogen which is used in the bio synthesis of protein (Campbell et al. 1967). When it comes to human beings, the role played by the L-aspartate is that it acts as a precursor of ornithine in the urea cycle. It also helps in the transmination reactions which produce oxaloaxetate in the gluconeogenic pathway thus resulting in the production of glucose (Narayan et al. 2007).
Interest in asparaginases was spurred due to their antitumor activity. There was a significant discovery that there was an association between anti-leukemic activity of guinea pig serum and its L-asparaginase activity together with the separation of the enzyme from Escherichia coli (Broome, 1961; Colboum et al. 2007). This discovery increased the attention and focus on bacterial L-asparaginases and this resulted in the identification and discovery of related enzymes present in other sources. Two of the bacteria enzymes (periplasmic proteins which are also referred to as type II asparaginases, from Escherichia coli (EcAII) and Erwinia chrysanthemi (ErA), have been used in the treatment of tumors and acute lymphoblastic leukemia for the last three decades (Byers, 2000).
Type 11 asparaginases enzymes contain two conserved amino-acid motifs.
Two amino-acid motifs conserved in bacterial asparaginases:
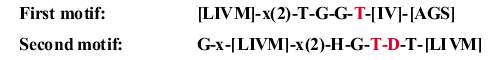
These types of asparaginases are homo-tetramers and, they have four active sites. Each of these active sites is as a result of amino acids produced by the two monomers, including the amino acids released by the conserved motifs.
In the nomenclature of EcA11, there are four subunits which are labeled ABCD while the active site competent intimate dimers have two subunits; AC and BD. The mechanism of the asparaginase reaction is the variant of the reaction which is catalyzed by serine proteases (Nawaz et al. 1998). However, the function of the nucleophilic serine is played by threonine (12 or 90 in EcAII sequence)
Asparaginases have an anti-tumor activity which is as a result of their high affinity for the substrate, L-asparagine. Because of the depletion of L-asparagine in the circulating pools, this results to starvation of the tumor cells. These cells have low levels of L-asparagine synthesis (Oettgen et al. 1967). The hydrolysis of L-glutamine is also possible especially by enzymes which have asparaginase activity. In instances where the better best is L-glutamine, the enzymes are referred to as glutaminase-asparaginases (EC 3.5.1.38). Glutaminase-asparaginases have similar tertiary and quaternary structure as those of type 11 asparaginases. The E. coligenome also encodes a related, cytoplasmic (type I) asparaginase (EcAI) (Karlson et al. 1993).
There are several uses of the asparaginases enzymes. They are used in industrial activities and, in pharmaceutical purposes as well.
Medical purposes of asparaginases enzymes: E-coli strains constitute the major source of medical asparaginase. Some of them include Asparaginase Medac, Ciderolase, and Oncaspar. There is a new recombinant E. coli asparaginase called Spectrila which has been developed (Barton et al. 1991; Marini et al. 1987). Asparaginase enzymes can be given through injections or as subcutaneous and intramuscular injections. One major advantage is that they do not cause any tissue irritation.
Food manufacturing: Asparaginases are used as processing aids in the manufacture and processing of food. They are used so that they can prevent the formation of acrylamide. This is a carcinogen that is present in most starchy food products especially snack (Barterlink et al. 2006). Acrylamide which is a carcinogen is produced when starchy foods are being prepared. Starchy foods naturally have amino acid asparagine, when they are heated; the asparagine goes through a process referred to as the Maillard reaction. This process is responsible for the brown color in fried foods. It is responsible for the toasted flavor and their crust (Amena et al. 2010; Barterlink et al. 2006). When the Maillard process is taking place, there are some carcinogens which are produced (such as acrylamide and some heterocyclic amines).
When asparaginase is added prior to cooking any starchy foods, asparagine is transformed into ammonium and aspartic acid, which is also another amino acid. Consequently, this ensures that asparagine does not part in the Maillard reaction. When asparagine takes part in the Maillard reaction, it results in the production of acrylamide; therefore, the absence of asparagine plays a role in hindering the formation of acrylamide (Boyse et al 1976). It is not possible to entirely eliminate acrylamide because of the pathway formations of some asparagine-independent components. The use of asparaginases reduces and lowers the level of acrylamide present in starchy foods. It does not alter with the appearance or the taste or flavor of the food.
In a study conducted by Erima et al., the results showed that some bacteria and fungi possess the ability to produce extracellular Asparaginase activity. It is therefore possible to isolate the active enzyme from culture filtrates. The basis of the clinical activity of Asparaginase is due to the ability of reducing circulating L-asparagine in the blood system (Campbell et al. 1967; Narayana et al. 2007). There are some neoplastic cells which are dependent on extracellular supplies of this amino acid, therefore, when they are deprived of L-asparagine, they die.
Some vaccine preparations have high L-asparaginase related activity and as such, the enzyme has been used in the potency control of such activities especially in the manufacturing process (Beumer-Jachmans, M. P. 1973). Some of the vaccines used in the immunotherapy of a variety of cancers, for example, breast cancer and lymphoblastic leukemia have therapeutic repercussions and this has been linked to the activities of the asparaginase. L-asparaginase includes all the enzymes which utilize and use the L-asparagine as a substrate (Amena et al. 2010). The hydrolysis of L-asparagine and L-glutamine is catalyzed efficiently with glutaminase asparaginases. These are enzymes which belong to the second class. Enzymes with asparaginase activity play a crucial role in the metabolism of living things and in pharmacology as well.
There are several side effects of asparaginase enzymes. First and foremost, it causes a hypersensitivity reaction and allergy as well. It can also result in anaphylaxis. Because it reduces the synthesis of protein, it can lead to coagulopathy. It reduces the synthesis of coagulation factors and anticoagulant factors (protein C and antithrombin III) and these enzymes are responsible for preventing bleeding and thrombotic events in case of a stroke (Campbell et al. 1967; Narayana et al. 2007). Though not in serious levels, asparaginase leads to the suppression of the bone marrow. Pancreatitis is also one of the side effects of the enzyme.
Conclusion
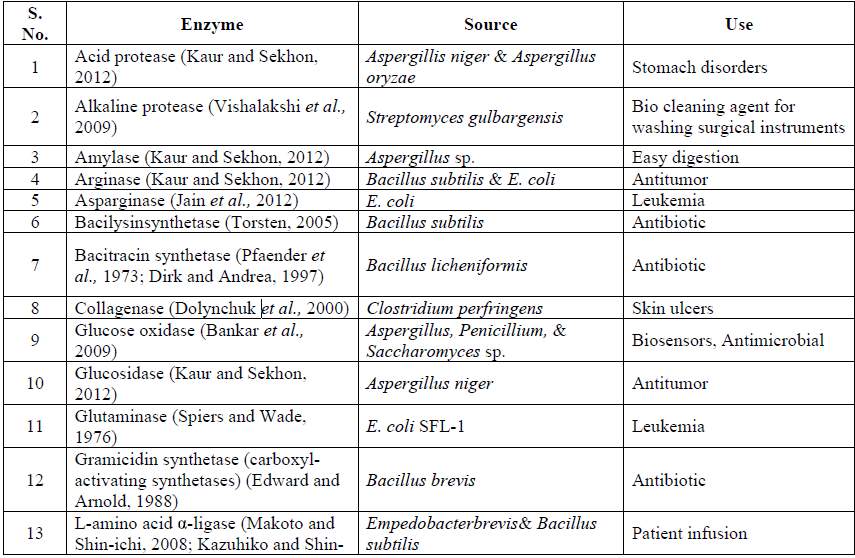
The use of microbial enzymes has increased in the last decade. Enzymes are now being used for clinical test reagents and there is a growing market for therapeutic enzymes. Most therapeutic enzymes are available in pills, capsules and food supplements. Medically important enzymes produced by microorganisms are applied as anti0ibflammatories, antimicrobial, anticoagulants, thrombolytic and digestive aids. These enzymes can also be used to treat diseases which have become resistant to drugs or antibodies. The use of enzymes and drugs in the treatment of disease can induce synergistic impacts and therefore treat several diseases through counteracting their side effects.
Table.4 Microbial therapeutic enzymes and their application
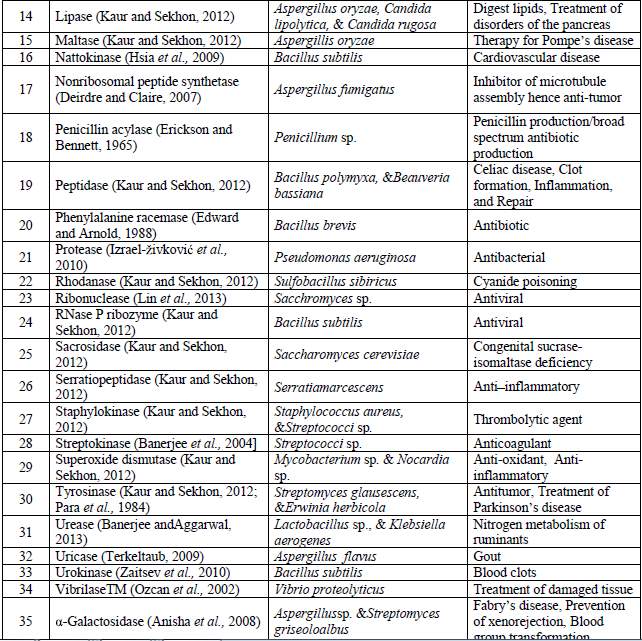
References
Amena S, Vishalakshi N, Prabhakar M, Dayanand A, Lingappa K: Production, purification and characterization of l-asparaginase from Streptomyces gulbargensis. Brazilian Journal of Microbiology,2010;41(1): 173-178
Basha NS, Rekha R, Komala M, Ruby S: Production of extracellular Anti-leukaemic enzyme L asparaginase from Marine Actinomycetes by solid state and submerged fermentation: Purification and characterization. Tropical Journal of Pharmaceutical Research, 2009; 8 (4): 353-360.
Boyse EA, Old LJ,Campbell HA, Mashburn LT: Suppression of murine leukemia’s by L-asparaginase. Incidence of sensitivity among leukemias of various types: Comparative inhibitory activities of guinea pig serum L-asparaginase and Escherichia coli Lasparagianse. Journal of Experimental Medicine, 1967; 125(1):17-31.
Kerem, B., J. M. Rommens, J. A. Buchanan, D. Markiewicz, T. K. Cox, A. Chakravarti, M. Buchwald, and L. C. Tsui. 1989. Identification of the cystic fibrosis gene: genetic analysis. Science 245:1073-1080.
Cystic Fibrosis Genetic Consortium. Cystic Fibrosis Mutation Database. 2009.
Tsui, L. C. 1992. The spectrum of cystic fibrosis mutations. Trends Genet 8:392-398.
Kerem, E., M. Corey, B. Kerem, J. M. Rommens, D. Markiewicz, H. Levison, L. C. Tsui, and P. Durie. 1990. The relation between genotype and phenotype in cystic fibrosis- analysis of the most common mutation (deltaF508). N.Engl.J.Med. 323:1517-1522.
The Cystic Fibrosis Genotype-Phenotype Consortium. 1993. Correlation between genotype and phenotype in patients with cystic fibrosis. N.Engl.J.Med. 329:1308-1313.
Campbell HA, Mashburn LT, Boyse EA, Old LJ: Two Lasparaginase from Escherichia–coli B. Their separation, purification and antitumor activity. Biochemistry, 1967; 6(3):721-730.
Randell SH, Boucher RC. Eff ective mucus clearance is essential for respiratory health. Am J Respir Cell Mol Biol 2006;35(1):20-28.
Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest 2002;109(5):571-577.
Wong JY, Rutman A, O’Callaghan C. Recovery of the ciliated epithelium following acute bronchiolitis in infancy. Thorax 2005;60(7):582-587.
Carroll NG, Mutavdzic S, James AL. Increased mast cells and neutrophils in submucosal mucous glands and mucus plugging in patients with asthma. Thorax 2002;57(8):677-682.
Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. Recombinant human DNase I reduces the viscosity of cystic fi brosis sputum. Proc Natl Acad Sci U S A 1990;87(23):9188-9192.
Riordan, J.R., et al., Identification of the cystic fibrosis gene: cloning and
characterization of complementary DNA. Science, 1989. 245(4922): p. 1066-73.
Gadsby, D.C., P. Vergani, and L. Csanady, The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature, 2006. 440(7083): p. 477-483.
Hwang, T.C. and K.L. Kirk, The CFTR ion channel: gating, regulation, and anion permeation. Cold Spring Harb Perspect Med, 2013. 3(1): p. a009498.
Derichs, N., Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Eur Respir Rev, 2013. 22(127): p. 58-65.
Cheung, J.C. and C.M. Deber, Misfolding of the cystic fibrosis transmembrane conductance regulator and disease. Biochemistry, 2008. 47(6): p. 1465-73
Andersen, D.H., Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathologic study. American Journal of Diseases of Children, 1938. 56(2): p.344-399.
Ramjeesingh, M., et al., Dimeric cystic fibrosis transmembrane conductance regulator exists in the plasma membrane. Biochem J, 2003. 374(Pt 3): p. 793-7.
Riordan, J.R., CFTR function and prospects for therapy. Annu Rev Biochem, 2008. 77: p. 701 26.
Shah PL, Scott SF, Knight RA, Marriott C, Ranasinha C, Hodson ME. In vivo eff ects of recombinant human DNase I on sputum in patients with cystic fi brosis. Thorax 1996;51(2):119-125.
Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME. Eff ect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fi brosis. The Pulmozyme Study Group. N Engl J Med 1994;331(10):637-642.
Greally P. Human recombinant DNase for mucus plugging in status asthmaticus. Lancet 1995;346(8987):1423-1424.
Puterman AS, Weinberg EG. rhDNase in acute asthma. Pediatr Pulmonol 1997;23(4):316-317.
Durward A, Forte V, Shemie SD. Resolution of mucus plugging and atelectasis after intratracheal rhDNase therapy in a mechanically ventilated child with refractory status asthmaticus. Crit Care Med 2000;28(2):560-562.
Patel A, Harrison E, Durward A, Murdoch IA. Intratracheal recombinant human deoxyribonuclease in acute life-threatening asthma refractory to conventional treatment. Br J Anaesth 2000;84(4):505- 507.
Narayana KJP, Kumar KG, Vijayalakshmi M: Lasparaginase production by Streptomyces albidoflavus. Indian Journal of Microbiology, 2007; 48(3): 331-336.
Oefner, C. & Suck, D. (1986) J. Mol. Biol. 192, 605-632.
Funakoshi, A., Tsubota, Y., Wakasugi, H., Ibayashi, H. & Takagi, Y. (1977) J. Biochem. 82, 1771-1777.
Murai, K., Yamanaka, M., Akagi, K., Anai, M., Mukai, T. & Omae, T. (1978) Biochim. Biophys. Acta 517, 186-194.
Byers, HL. (2000) Isolation and characterisation of sialidase from a strain of Streptococcus oralis. J Med Microbiology, 49:235–244.
Colbourn T, Asseburg C, Bojke L, Philips Z, Welton N, Claxton K, Ades A, Gilbert R. (2007) Preventive strategies for group B Streptococcal and other bacterial infections in early infancy: cost effectiveness and value of information analyses. BMJ, 2-7.
Gonzalez SN, Cardozo R, Apella MC, Oliver G. (1995) Biotherapeutic role of fermented milk. Biotheraphy, 8:126-134.
Macfarlane GT, Gibson GR. (1994) Metabolic activites of the normal colonic flora. Springer-Verlag, 17-52.
Love, J. D. & Hewitt, R. R. (1979) J. Biol. Chem. 254, 12588- 12594.
Ito, K., Minamiura, N. & Yamamoto, T. J. (1984) Biochemistry 95, 1399-1406.
Nawaz MS, Zhang D, Khan AA, Cerniglia CE: Isolation and characterization of Enterobacter cloacae capable of metabolizing asparagine. Applied Microbiology & Biotechnology, 1998; 50(5): 568-572.
Du, K., M. Sharma, and G.L. Lukacs, The [Delta]F508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat StructMol Biol, 2005. 12(1): p. 17-25.
Cheng, S.H., et al., Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell, 1990. 63(4): p. 827-34.
Sherman JM. Bronchial lavage in patients with cystic fi brosis: a critical review of current knowledge. Pediatr Pulmonol 1986;2(4):244-246.
Connett GJ, Doull IJ, Keeping K, Warner JO. Flexible fi bre-optic bronchoscopy in the management of lung complications in cystic fi brosis. Acta Paediatr 1996;85(6):675-678.
Slattery DM, Waltz DA, Denham B, O’Mahony M, Greally P. Bronchoscopically administered recombinant human DNase for lobar atelectasis in cystic fi brosis. Pediatr Pulmonol 2001;31(5):383-388.
de Blic J, Midulla F, Barbato A, Clement A, Dab I, Eber E, Green C, Grigg J, Kotecha S, Kurland G, Pohunek P, Ratjen F, Rossi G. Bronchoalveolar lavage in children. ERS Task Force on bronchoalveolar lavage in children. European Respiratory Society. Eur Respir J 2000;15(1):217-231.
Tannenbaum E, Prasad SA, Dinwiddie R, Main E. Chest physiotherapy during anesthesia for children with cystic fi brosis: Eff ects on respiratory function. Pediatr Pulmonol 2007;42(12):1152-1158.
Price JF. The need to avoid general anaesthesia in cystic fi brosis. J R Soc Med 1986;79 Suppl 12:10- 12.
Aitken ML, Burke W, McDonald G, Shak S, Montgomery AB, Smith A. Recombinant human DNase inhalation in normal subjects and patients with cystic fi brosis. A phase 1 study. JAMA 1992;267(14):1947-1951.
Denning, G.M., et al., Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature, 1992. 358(6389): p. 761-764.
Qu, B.H. and P.J. Thomas, Alteration of the cystic fibrosis transmembrane conductance regulator folding pathway. J Biol Chem, 1996. 271(13): p. 7261-4.
Lukacs, G.L. and A.S. Verkman, CFTR: folding, misfolding and correcting the _F508 conformational defect. Trends in Molecular Medicine, 2012. 18(2): p. 81-91.
Castaldo, G., et al., Severe liver impairment in a cystic fibrosis-affected child homozygous for the G542X mutation. American Journal of Medical Genetics, 1997.69(2): p. 155-158.
Bompadre, S.G., et al., G551D and G1349D, Two CF-associated Mutations in the Signature Sequences of CFTR, Exhibit Distinct Gating Defects. J Gen Physiol, 2007.129(4): p. 285-298.
Oettgen HF, Old LJ, Boyse EA, Campbel HA, Philips FS, Clarkson BD, Tallol L, Leeper RD, Schiwartz MK, Kim JH: Inhibition of leukemias in man by Lasparaginase. Cancer Research, 1967;27(12):2619-2631.
Olempska-Beer ZS, Merker RI, Ditto MD, DiNovi MJ: Food-processing enzymes from recombinant microorganisms – a review. Regulatory Toxicology and Pharmacology, 2006; 45:144-158
Karlson KH, Jr., Pickert CB, Schexnayder SM, Heulitt MJ. Flexible fi beroptic bronchoscopy in children on extracorporeal membrane oxygenation. Pediatr Pulmonol 1993;16(4):215-218.
Nussbaum E. Pediatric fl exible bronchoscopy and its application in infantile atelectasis. Clin Pediatr (Phila) 1985;24(7):379-382.
Wong KS, Lin TY, Lan RS. Evaluation of chronic atelectasis in children using chest computed tomography and bronchoscopy. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi 1996;37(3):193- 196.
Marini A, Franzetti M, Gios G, Flauto U, Arosio A, Maccabruni M, Rondini G, Chirico G, Giancola A, Console V, et al. Ambroxol in the treatment of idiopathic respiratory distress syndrome. An interim report on a controlled double-blind multicenter study versus placebo. Respiration 1987;51 Suppl 1:60-67.
Barker AF. Bronchiectasis. N Engl J Med 2002;346(18):1383-1393.
Ahmad I, Nemet D, Eliakim A, Koeppel R, Grochow D, Coussens M, Gallitto S, Rich J, Pontello A, Leu S-Y et al (2010) Body composition and its components in preterm and term newborns: a cross-sectional, multimodal investigation. Am J Hum Biol 22:69–75
Alcorn J, McNamara PJ (2003) Pharmacokinetics in the newborn. Adv Drug Deliv Rev 55:667–686
Atkinson M, Budge H (2011) Review of the NICE guidance on neonatal jaundice. Arch Dis Child Educ Pract Ed 96:136–140
Barrett DA, Rutter N (1994) Transdermal delivery and the premature neonate. Crit Rev Ther Drug Carrier Syst 11:1–30
Bartelink IH, Rademaker CMA, Schobben AFAM, van den Anker JN (2006) Guidelines on paediatric dosing on the basis of developmental physiology and pharmacokinetic considerations. Clin Pharmacokinet 45:1077–1097
Berseth CL (1996) Gastrointestinal motility in the neonate. Clin Perinatol 23:179–190
Blake MJ, Gaedigk A, Pearce RE, Bomgaars LR, Christensen ML, Stowe C, James LP, Wilson JT, Kearns GL, Leeder JS (2007) Ontogeny of dextromethorphan O- and N-demethylation in the fi rst year of life. Clin Pharmacol Ther 81:510–516
Schibli S, Durie PR, Tullis ED: Proper usage of pancreatic enzymes. Curr Opin Pulm Med 2002, 8 :542-546.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biomedical Science"
Biomedical Science focuses on how cells, organs and systems function in the human body and underpins much of modern medicine. Biomedical Science applies parts of natural and/or formal sciences to help develop advances in healthcare.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: