Dissertation on Amino Acids Role in Appetite Regulation
Info: 10152 words (41 pages) Dissertation
Published: 12th Nov 2021
Investigating the downstream intracellular signalling of NmU and NmS on the NMU2 receptor
Abstract
The Neuromedin U type 2 receptor (NMU2) is currently classed as a family A G-protein coupled receptor (GPCR). NMU2 is activated by the endogenous peptides Neuromedin U (NmU) and Neuromedin S (NmS). NMU2 activation by both NmU and NmS has been implicated to have anorexigenic effects in various studies. Currently conflicting roles of leptin acting on the hypothalamic-pituitary-adrenal (HPA) axis following NMU2 activation means the pathway which NMU2 follows to regulate the feeding and appetite is currently unclear. Expanding our understanding of NmU and NmS mediated NMU2 intracellular signalling mechanisms, provides the potential to understand the role of NMU2 in feeding regulation. NmS is shown to have a slightly higher potency to NMU2 compared to NmU. Therefore, our investigation focused on identifying the cause of this. As NMU2 is Gαq coupled, initially the change in [Ca2+]I caused by NmU and NmS activation of NMU2 was compared. This showed no significant difference. Therefore, the activation of Erk1/2 caused by NmU and NmS was compared. The time course activation of Erk1/2 by NmS was shown to be greater than NmU, suggesting NmU and NmS activate Erk1/2 via different pathways. To identify if Erk1/2 was activated by G-protein dependent or G-protein independent pathways, a pH2 acid wash western blot protocol was used to remove extracellular and ligand bound to NMU2 and to record the level of activated pErk1/2. In summary, both NmU and NmS show G-protein dependent activation of Erk1/2. However, NmU shows both G-protein dependent and independent activation of Erk1/2.
Introduction
Neuromedin U (NmU) and Neuromedin S (NmS) are endogenous ligands belonging to the Neuromedin family of peptides.
Two major forms of NmU exist. The 23 amino acid NmU-23 and 25 amino acid NmU-25 are the long forms. Also truncated 8 (NmU-8) and 9 (NmU-9) amino acid length forms have been isolated. NmU-25 and NmU-8 were isolated first from porcine spinal cord due to their ability to contract rat uterus, hence the suffix U (Hosoya et al., 2000). Recently novel NmU splice variants have been identified from goldfish gut and brain (NmU-21, NmU-25 and NmU-38) (Maruyama et al., 2008) and also the Chinese red belly toad (NmU-17) (Lee et al., 2005). Therefore, NmU has been identified in various organisms. Only NmU-25 is shown to be present from human cDNA and is located at chromosome 4q12 (Austin et al.,1995). The truncated NmU peptides are spice variants of the long forms and all NmU variants have five conserved amino acid residues on the C-terminal end of the peptide (-Phe-Arg-Pro-Arg-Asn-NH2) (Austin et al.,1995; Lee et al., 2005). The only exceptions are goldfish NmU-25 and NmU-21. The conserved amino acids are essential for activity of NmU. Substitution of amino acids in the conserved region reduces the ability of NmU to contract muscle (Hashimoto et al., 1991; Kurosawa et al., 1996). The N-terminus was also shown to be essential to NmU activity in amino acid substitution experiments (Kurosawa et al., 1996).
NmS is a 36 amino acid neuropeptide which also shows the same five conserved amino acid residues as NmU. NmS was isolated from the superchiasmatic nucleus of rat hypothalamus and is part of the Neuromedin family of peptides (Mori et al., 2005). NmS gene is located on chromosome 2q11.2 which differs from NmU (Mitchell et al., 2009). Despite this NmU and NmS have shown to have similar physiological roles in regulating contraction. NmU and NmS are both circulating hormones (Mitchell et al., 2009; Mitchell et al 2009a).
The conserved amino acids common to NmU and NmS underlie the ability of both agonists to bind the two Neuromedin receptors. First radiolabelled NmU was shown to bind the Neuromedin U type 1 receptor (NMU1) (Nandha et al., 1993). Subsequently several groups showed NmU to be the potent ligand for NMU1 (Fujii et al., 2000; Raddatz et al., 2000; Brighton et al., 2004). The gene encoding the 403 amino acid human NMU1 is located on chromosome 2q34-q37. The 406 and 402 amino acid variants of NMU1 are found in mice (Raddatz et al., 2000). However, due to varying locations of the initiation codon from NMU1 the exact amino acid length may vary. NMU1 was classified as a G-protein coupled receptor (GPCR) due to the reduced binding of ligands in the presence of a non-hydrolysable GTP substrate (Nandha et al., 1993). Soon after the discovery of NMU1, a second receptor which NmU binds was identified, known as Neuromedin U type 2 receptor (NMU2) (Fujii et al., 2000; Raddatz et al., 2000; Brighton et al., 2004). NMU2 shares a 51% protein homology to NMU1 (Shan et al., 2000). This level of homology coupled with conserved exon-intron boundaries suggests NMU1 and NMU2 may be products of a duplication event (Shan et al., 2000). The gene encoding the 412 amino acid human NMU2 is located on chromosome 5q33.1. NMU1 and NMU2 show up to 75 and 817 sequence similarities in mice and rats respectively. However, due to varying locations of the initiation codon from NMU1 the exact amino acid length may vary. (Shan et al., 2000). Thus different length human NMU1 and NMU2 can occur, though both are GPCR’s (Raddatz et al., 2000).
The distribution of NMU1 and NMU2 spans several locations. Identification of NMU1 and NMU2 in the human body utilizes several techniques including northern blots, dot blots reverse transcriptase-polymerase chain reaction (RT-PCR) and in situ hybridization (Hosoya et al., 2000). Hence identifying protein expression levels underlies these techniques. NMU1 is predominantly expressed in peripheral tissues such as the small intestines, stomach testis lungs, kidneys, pancreas, uterus and adrenal gland (Hosoya et al., 2000). The presence of NMU1 in the brain has shown contrasting evidence. Hedrick (2000) fail to show the presence of NMU1 mRNA in the brain. However, Howard (2000) show NMU1 is present in the brain. Nonetheless, the levels of NMU1 in the brain is substantially lower than its presence in the periphery tissues (Hosoya et al., 2000). Conversely, NMU2 is present predominantly in the central nervous system of humans (Brighton et al., 2004). NMU2 is highly abundant in the hippocampus, medulla oblongata, thalamus and spinal cord. NMU2 is also present in lower amounts in peripheral tissues (Hosoya et al., 2000).
NMU1 and NMU2 both signal as family A GPCR. This is due NMU1 and NMU2 possessing conserved amino acid triplets (D/ERY motif) for ligand binding and G-protein coupling (Brighton et al., 2004; Rhee et al., 2000; Scheer, et al., 2000). Also they both have characteristically located cysteine residues in extracellular loops 2 and 3 (Brighton et al., 2004). These features of NMU1 and NMU2 are characteristic of family 1 GPCR’s. NMU2 is shown to exhibit Gαq and Gαi properties (Brighton et al., 2004). Upon investigations into other potential ligands for NMU1 only NmU and NmS are shown to increase [Ca2+]I and inositol phosphate (IP3) levels. Currently only the various forms of NmU and NmS were found to be an agonist of NMU1. However, NmS (IC50=1.0 × 10−10M) is shown to be slightly more potent to NMU2 in the CNS than NmU (IC50 = 6.8 × 10−10 M) (Mori et al., 2005).
NmU and NmS binding NMU1 and NMU2 has various roles throughout the human body. This is to be expected due to the widespread distribution of NMU1 and NMU2. Currently research is focused on the role of NmU in smooth muscle contraction and regulating feeding (Mitchell et al., 2009).
The effect of NmU in causing contraction of smooth muscle is shown to be species specific. Murine models show NmU enhances muscle contraction indirectly in the colon through effecting electrical activity controlling contraction by reducing the interval between successive contractions (Dass et al., 2007). This was shown to be limited to NMU1. Also NmU was shown to mediate contraction of mouse gallbladder and lower esophageal sphincter (Dass et al., 2007). However, in humans NmU is shown to directly affect blood flow by causing smooth muscle contraction upon stimulation of both NMU1 and NMU2 (Jones et al., 2006). Knockout studies show NmU binds NMU1 in gallbladder contraction (Jones et al., 2006). Conversely, the action of NmU in mediating murine uterus and vas deferens contraction occurs via NMU2. This shows the receptor which NmU binds is dependent on the location of action of NmU.
The central effects of NmU are shown as intracerebroventricular (i.c.v) injection of NmU reduces appetite and feeding. RT-PCR shows NmU is expressed centrally in in various murine, quail and goldfish models (Howard et al., 2000; Wren et al., 2002). All species show reduced appetite and increased food intake after i.c.v injection of NmU.
Originally, NmU is shown to inhibit feeding as i.c.v injection of NmU at the paraventricular nucleus (PVN) shows a dose dependent inhibition of food intake by acting on the hypothalamic-pituitary-adrenal (HPA) axis to cause NmU mediated increases in corticotropin-releasing hormone (CRH). This resulted in a leptin regulated increase in adrenocorticotropic hormone (ACTH) and corticosterone, which was shown to decrease when NmU is blocked using an anti-NmU IgG (Heiman et al., 1997; Van Dijk et al., 1997; Jethwa et al., 2006).
The effects of NmU in regulating appetite are believed to be occur via NMU2. This is due to the expression of NMU2 in the wall of the 3rd lateral ventricle of the hypothalamus (Hosoya et al., 2000). The NMU2 agonist EUK2010 decreased food intake and reduced body weight of mice. Treatment with EUK2010 showed increases in the appetite regulating hormones CRH and ACTH. These hormones are controlled by the leptin receptor. Hence originally NmU was believed to exerted its anorexigenic effects downstream of the leptin pathway (Fang et al., 2006).
However, contrasting evidence was identified as mice lacking leptin expression still showed weight less upon i.c.v injection of NmU. Also injection of leptin to mice failed to alter the expression of NmU gene in the hypothalamus. This shows a leptin-independent nature of NmU (Hanada et al., 2004). Also, NmU KO mice showed elevation of CRH but no change in ACTH levels (Thompson et al., 2004). NmU is shown to act in part on CRH as CRH KO mice fail to show reduced feeding (Hanada et al., 2004; Martinez et al., 2015). Therefore, the mechanism by which leptin acts on the HPA axis is unclear. However, despite the lack of a clear pathway, it is clear NmU is shown to reduce feeding and appetite.
Peier (2009) show the reduced feeding effects of NmU and NmS upon binding NMU2 are abolished in NMU2 KO mice. This is shown following both acute and chronic administration of NmU. More selective KO of PVN NMU2 in rats shows no significant effect on feeding but does show an increased preference of rats to high fat foods (Benzon et al., 2014). Therefore, the exact mechanism of NMU2 in feeding is currently unclear. To understand the role of NMU2 in regulating feeding and appetite, the intracellular signalling of NMU2 following stimulation by NmU or NmS must be understood.
NmU and NmS function via the NMU2 receptor. NmS shows a slightly higher potency to NMU2 compared to NmU (Mori et al., 2005). Therefore, our investigation aimed to identify if this difference in affinity of NmS, causes a different intracellular signalling pathway to be followed. To investigate this initially intracellular calcium levels were recorded from NmU and NmS on NMU2 in the presence of an acid wash protocol. The calcium levels were recorded due to the Gαq nature of NMU2. Gαq signalling also results in the activation of mitogen activated protein kinases (MAPK) such as extracellular regulated kinase (Erk1/2). Hence the level of Erk1/2 activation by NMU2 with NmU and NmS respectively was also compared to identify if Erk1/2 is activated by G-protein dependent or G-protein independent pathways. Any potential differences suggested from this data can explain the evolutionary requirement of two ligands, NmU and NmS, binding to the same NMU2 receptor to mediate appetite and feeding. Subsequently, clarification of NMU2 intracellular signalling could allow the potential for NMU2 to be pharmacologically targeted to treat feeding disorders such as obesity (Peier et al., 2009; Micewicz et al., 2015).
Methods
Cell culture
NMU2 stably expressed in human embryonic stem kidney cells (HEK293-NMU2) and cultured in flasks were examined for confluency and contamination. Cells were washed with 5ml Dulbecco’s phosphate-buffered saline (DPBS). 3ml Trypsin-Ethylenediaminetetraacetic acid (EDTA) (0.04% EDTA & 0.05%trypsin) solution was used to remove the cells from the flask surface. After detachment of cells 5ml growth media was added and the cells were re-suspended. The cells were then centrifuged at high speed for 4 min. the supernatant was poured away and the pellet was re-suspended in 5ml growth media and transferred to a flask for incubation at 37°C, 95% humidified air and 5% CO2 until the cells were confluent for use.
Seeding and calcium signalling
To conduct calcium recordings 96 and 24 well/plates were covered with 100uL or 500μL poly-D-lysine hydrobromide respectively and incubated for 30 min at 4 °C. The poly-D-lysine hydrobromide was then removed and each well was washed with 200μL DPBS. 100μL of cell solution from the incubated flask was seeded into the wells and cultured for 24 hours to create a monolayer of cells. Then the growth media was removed and cells were washed with KHB (4.7mM KCl, 118mM NaCl, 11.7mM glucose, 1.2mM MgSO4, 4.2 mM NaHCO3, 1.2mM KH2PO4 and 1.3mM CaCl2, pH 7.4, BSA (0.1%)) solution. Each well was loaded with fluo-4-acetoxymethyl ester (fluo-4-AM) solution (2μL fluo-4-AM + 1ml KHB) and incubated at 37°C, 95% humidified air and 5% CO2 for 45 min. The fluo-4-AM was then removed and each well was washed with KHB and then KHB was added to each well after the final wash. Fluo-4-AM is a calcium sensitive dye used to measure changes in free intracellular calcium levels by increasing fluorescence when bound to calcium. The change in fluorescence was recorded by the NOVOstar plate-reader at an excitation wavelength of 488nm and a detection of 510nm. The change in florescence was recorded from monolayers of cells containing NMU2 stimulated by NmU and NmS at various concentrations and was used as a representation of [Ca2+]I to construct a concentration response curve. The data was plotted using GraphPad Prism 6 Software.
Acid wash control
HEK293-NMU2 cells were seeded onto 96 well/plates and loaded with Fluor-4-AM as shown previously. Cells were exposed to a pH2 solution (KHB, 1M HCl) for 20 seconds, after which the pH2 solution was removed by aspiration. The cells were washed with 100uL KHB two times and 100uL KHB was added to each well after the final wash. This occurred before ligand stimulation by the NOVOstar plate-reader. The monolayers of HEK293-NMU2 cells were then stimulated with NmU or NmS and the change in fluorescence of Fluo-4-AM was recorded as a representation of changes in [Ca2+]I by the NOVOstar plate-reader (shown figure 1).
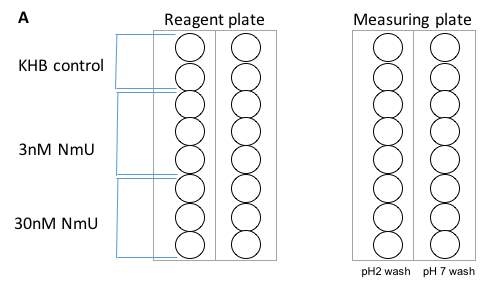
Figure 1: Each circle represents a well from the 96 well/plate. This shows the recoding and measuring plates used in the NOVOstar plate-reader. To the reagent plate 100μL KHB was added to the first two wells of each column as a control. The three wells bellow contained the 30nM NmU. The final three wells contained the 100mM NmU. The measuring plate contained cells loaded with Flou-4-AM and washed with either pH2 solution or KHB (pH7.4). The same protocol was repeated for NmS.
Challenge/re-challenge protocol
HEK293-NMU2 cells were seeded onto 96 well/plates and loaded with Fluor-4-AM as shown previously. 100uL 30nM concentrations of NmU and NmS were added for 1 min to each well to stimulate the HEK293-NMU2 cells (first stimulation). The ligands were then removed using aspiration. One column on the measuring plate was washed using 100uL pH2 solution (KHB, 1M HCl) for 20 seconds. The adjacent column was washed with 100uL KHB (pH7.4) for 20 seconds (shown figure 2). Then all wells were washed two times using KHB and KHB was added to the wells after the final wash. The change in fluorescence of Fluo-4-AM was recorded as a representation of changes in [Ca2+]I for each strip by the NOVOstar plate-reader.
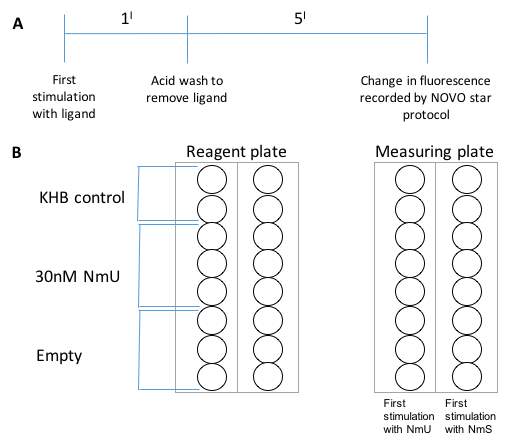
Figure 2: Each circle represents a well from the 96 well/plate. 2A shows the time course of events in the challenge/re-challenge protocol. 2B shows the measuring and reagent plate. To the reagent plate 100μL KHB was added to the first two wells of each column as a control. The three wells bellow contained the 30nM NmU. The final three wells were empty. The measuring plate contained cells loaded with Flou-4-AM and stimulated by NmU or NmS.
Western blotting
Western blot was conducted to record changes in the phosphorylation of extracellular signal-regulated kinase 1/2 (Erk1/2). 24 well/plates coated with 200μL poly-D-lysine hydrobromide were seeded with HEK293-NMU2 cells. 200uL starvation media was added and cells were starved for a minimum of 18 hours. After starvation media was replaced with 200uL KHB.
Time course western blot cell stimulation
Each well was stimulated with 200uL 30nM NmU or NmS for various durations between 0-120 min.
Acid wash western blot cell stimulation
Monolayers of HEK293-NMU2 cells were stimulated with 200uL 30nM NmU or NmS for 1min (first stimulation). The addition of 200uL KHB to two wells was used as a control. The ligands were removed by aspiration. The cells were washed using 100uL pH2 solution (KHB, 1M HCl) for 20 seconds. Then the cells were washed with 200uL KHB and KHB was added to the wells after the final wash. Each well was left to recover for different durations between 0-120 min after wash with pH2 solution. To ensure all reactions end at the same time, first stimulation was conducted at the longest recovery time (120 min). Then the whole process was repeated using a first stimulation time of 5 min.
Sample preparation
Then the cells were washed two times with iced DPBS. The cells were then incubated on ice with 200uL sample buffer (125mM Tris-HCL, 10% SDS, 20% glycerol, 0.01% bromophenol blue, 250mM dithiothreitol) to lyse the cells. The lysates were then transferred to a microfuge tube and centrifuged at high speed for 1 min. then the lysates were placed on a 100°C heat block for 5 min. The lysates would then be run on the gel.
Gel preparation
The 10ml solution for 10% resolving gel was made according to Sambrook (2006). The mini-BioRad electrophoresis system was used to generate a 1.5mm thick gel (Sambrook et al., 2006). The solution was left for 30 min to allow the gel to solidify. 1ml isopropanol was added to ensure the gel edge was flat. After 4ml of 10% stacking gel solution (acrylamide, 30% Tris-HCL, 10% APS, 1M pH 6.8 SDS, 10% TEMEDand topped up to 4ml with water) was added on-top of the solidified resolving gel and an 18-well comb was added to form wells. After 30 min the resolving gel solidified and the mini-BioRead gel was attached to an electrophoresis tank filled with running buffer (25mM Tris-HCL, 25mM glycine, 192mM SDS 0.1% pH8.6). Then 20uL of the lysate was loaded into the wells and electrophoresis occurred at 180V for 52 mins.
Semi-dry transfer
After washing semi-dry transfer was conducted using filter paper and membrane which was cut to the measurements of to the gel. The membrane was activated by placing it in 100% methanol for 10 seconds, then washing in water for 2 mins. The filter paper and membrane were then stored in transfer buffer (48mM Tris, 40mM glycine, 0.0375% SDSand 20% methanol) for 10 mins. The papers and membrane were removed from the transfer buffer. Then the gel was placed on-top of the membrane with three filter papers above and below. Then the semi-dry transfer was conducted using BioRad Power Pac 300 at 15V for 30 min.
After the membrane was immersed with light rocking for one hour in 5ml Tris buffered saline with Tween 20 and 5% fat free milk (TBST). The membrane was then washed with TBST (without milk) three times for 1min with light rocking. The membrane was then folded into a Falcon tube and incubated at 4°C overnight with rolling, with the primary monoclonal antibody for Erk1/2 (phosphor-p44/42 mitogen activated protein kinase (MAPK)). After the membrane was washed with TBST (without milk) for 8mins three times. Then the membrane was incubated with anti-rabbit HRP-linked secondary antibody for one hour at room temperature with moderate shaking. Then the membrane was washed with TBST (without milk) for 8 mins three times.
UptiLightTM chemiluminescent reagent was used to detect the pErk1/2 activation. The exposure time of the membrane to the X-ray film was between 10-100 seconds. Image J software was sued to quantify the pErk1/2 signal by comparing the size of the pErk1/2 marker.
Results
Calcium signalling
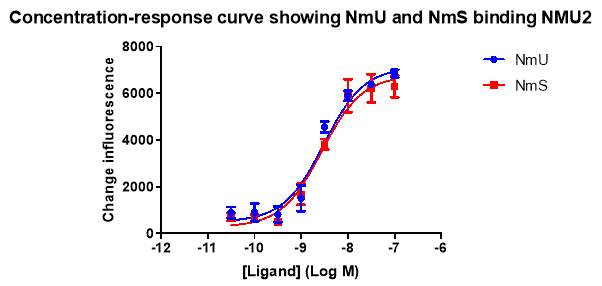
Figure 3: This shows the change in fluorescence as an indicator of intracellular calcium ([Ca2+]I) recorded using the NOVOstar plate reader. Monolayers of HEK293-NMU2 cells were stimulated with NmU (blue) or NmS (red) at different ligand concentrations. The change in fluorescence of Fluo-4-AM was recorded as a representation of changes in [Ca2+]I by the NOVOstar plate-reader. This experiment was conducted as part of N=5.
The concentration response curve above shows as each ligand is added a calcium response is elicited. The higher the concentration of ligand added results in a larger change in florescence hence a larger rise in calcium. The calcium response was low when either ligand was added at 0.1nM, 0.3nM and 1nM. The calcium response increases between 3nM to 10nM. Then the response plateaus between 30nm and 10nm. Overall the two curves show NmU and NmS elicit similar calcium responses. The concentration response curve was recorded to identify the EC50 values for NmU and NmS. The recorded EC50 value for NmU is shown as 3.03 +- 0.0468 (nM). The NmS EC50 is 2.77 +- 0.049074773 (nM).
Acid wash control
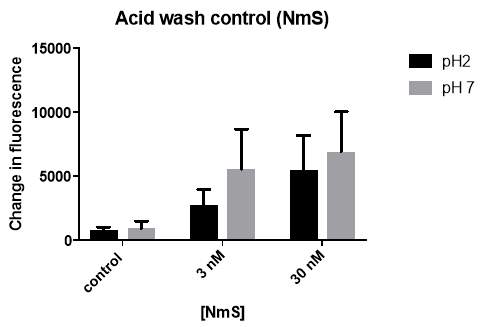
Figure 4: Monolayers of HEK293-NMU2 cells were washed with either pH2 acid (3nM and 30nM) or pH7.4 KHB (control). After a 5 min recovery period the monolayers of HEK293-NMU2 cells were then stimulated with NmS and the change in fluorescence of Fluo-4-AM was recorded as a representation of changes in [Ca2+]I by the NOVOstar plate-reader. This was conducted as part of N=3.
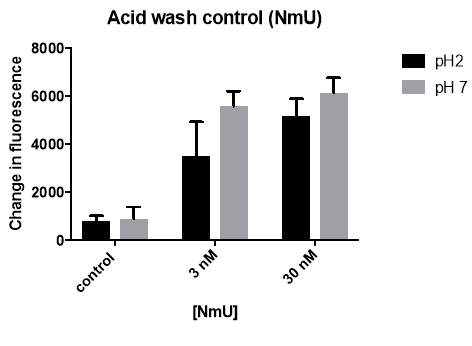
Figure 5: Monolayers of HEK293-NMU2 cells were washed with either pH2 acid (3nM and 30nM) or pH7.4 KHB (control). After a 5 min recovery period the monolayers of HEK293-NMU2 cells were then stimulated with NmU and the change in fluorescence of Fluo-4-AM was recorded as a representation of changes in [Ca2+]I by the NOVOstar plate-reader. This was conducted as part of N=3.
This was conducted as part of N=3.
This experiment was used identify if 20 second application of acid effects the cells ability to initiate a calcium response. The concentrations of 3nM and 30nM ligand were used as the previous experiment identified that a calcium response occurs at both concentrations. By comparing pH2 acid solution wash and pH7.4 KHB wash calcium responses when stimulated with 30nM NmS (figure 5), no significant difference (P value = 0.6379) is observed. Comparison of pH2 acid solution wash and pH7.4 KHB wash calcium responses when stimulated with 30nM NmS (figure 6), no significant difference (p value = 0.0740) is again observed. As a result, the acid wash was shown not to effect the cells calcium response to ligand at 30nM. This experiment showed the pH2 acid wash had no significant effect on the calcium response compared to the pH7.4 KHB wash. Hence the acid wash protocol was used in subsequent experiments to remove ligand bound to the receptor.
Challenge/re-challenge protocol
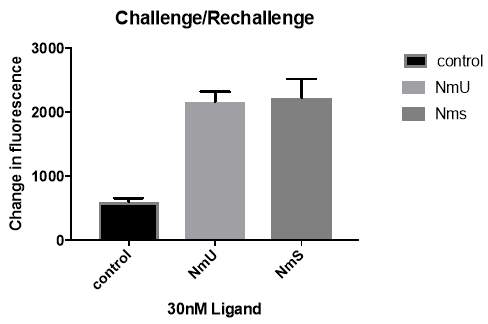
Figure 6: Monolayers of HEK293-NMU2 cells were first challenged with 30nM NmU or NmS. The cells were then washed using pH2 acid solution (NmU and NmS) or with pH 7.4 KHB (control). After a recovery period, the cells were re-challenged with 30nM NmU. The change in fluorescence following re-challenge, of Fluo-4-AM was recorded as a representation of changes in [Ca2+]I by the NOVOstar plate-reader. The control shows the calcium response when the first exposure is with KHB, no ligand. This experiment was conducted as part of N=3.
This experiment was conducted to investigate if the slightly higher potency of NmS to NmU causes NMU2 to change [Ca2+]I differently. The bar chart shows the change in [Ca2+]I recorded after first exposure to NmS elicits a slightly larger response compared to NmU. However, analysis shows no significant difference between the calcium responses from first exposure to NmU and NmS after removal by acid wash and challenged with NmU. This is supported by a p value of 0.3013. Also the calcium response was significantly greater than the control.
Time course western blot cell stimulation
The purpose of this experiment was to record changes in the phosphorylation of extracellular signal-regulated kinase 1/2 (pErk1/2) following activation by NmU or NmS for different durations. The level of pErk1/2 activation for NmS is greater at each time point compared to the pErk1/2 for NmU. Also, the levels of pErk1/2 are sustained from 5 min to 120 Min time points for both ligands.
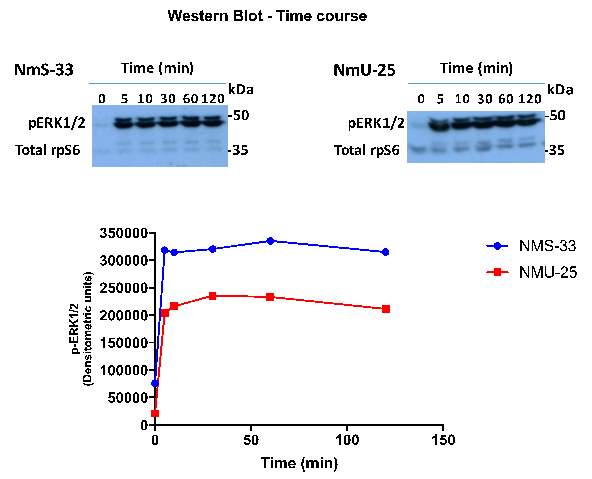
Figure 7: Monolayers of HEK-NMU2 cells cultured on 24 well plates. Each well was stimulated with 200uL 30nM NmU or NmS for various durations between 0-120 min.
The pErk1/2 response was recorded from a western blot. Each point on the graph for each ligand represents the time in which the ligand was left present to NMU2 (0, 5, 10, 30, 60 and 120 Min). This experiment was conducted as N=1.
Acid wash western blot cell stimulation
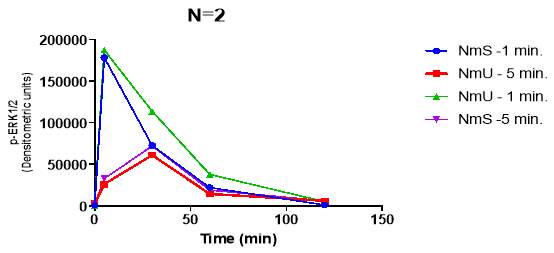
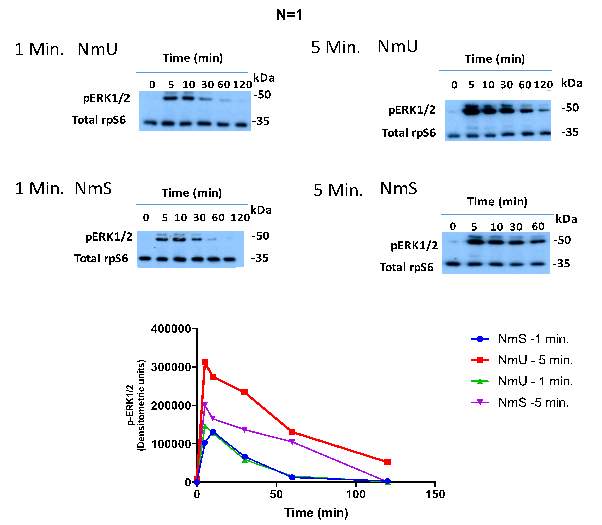
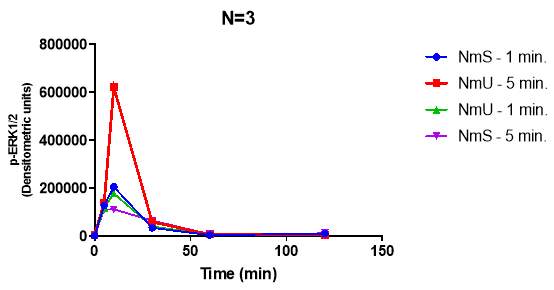
Figure 8: Monolayers of HEK293-NMU2 cells cultured on 24 well plates. They were stimulated with 30nM NmU (red and green) or NmS (blue and purple). Then the ligand was removed after either 1 min (blue and green) or 5 min (red and purple) using pH2 acid solution. The cells were washed using 100uL pH2 KHB for 20 seconds. Then the cells were washed with 200uL KHB and KHB was added to the wells after the final wash. Each well was left to recover for different durations between 0-120 min after wash with pH2 solution. The pErk1/2 activation was recorded for each ligand at each time point and plotted above. Each graph represents one repeat experiment. This investigation was conducted as N=3.
This experiment was conducted to identify if the if the slightly higher potency of NmS to NmU causes NMU2 to alter the levels of pErk1/2 differently for each ligand and to identify if NmU and NmS activate pErk1/2 in a G-protein independent manner. Overall the western blot acid wash investigation results are not comparable due to large variations in the data obtained for each repeat. Hence the statistical tests show no consistent conclusions between each replicate experiment. Despite this a key feature of all the experiments is the graded decrease in pErk1/2 response as the time points increase. When either NmU or NmS was first stimulated for 1 Min or 5 Mins, NMUR2 will be activated. As extracellular ligand and NMUR2 bound ligand is removed by acid wash, pErk1/2 responses decrease over time. This is shown as the 120 min time point of both ligands results in a lower pErk1/2 response compared to 5 min time point.
From N=1 the pErk1/2 response of NmU 5 min (red) is shown to be significantly greater than NmU 1 min (green) at the 5 min and 10 min time points. This is supported by a p value of 0.0269 and 0.0192 for the 5 min and 10 min time points respectively. Also NmU 5 min (red) is shown to be significantly greater than NmU 1 min (green) at the 10 min time point. This is supported by a p value of 0.0205.
Also, both NmU and NmS show pErk1/2 activation following the acid wash, suggesting both ligands can signal when no ligand is bound to the receptor.
Discussion
NMU2 is currently classed as a family A GPCR. NMU2 is a Gαq coupled GPCR, hence activation of NMU2 by NmU or NmS results in increases in intracellular calcium and Erk1/2 activation (Shenoy et al., 2011).
NmS is shown to be slightly more potent to NMU2 compared to NmU (Mori et al., 2005). This is supported by the EC50 values obtained from our calcium signalling investigation (figure 3). The challenge/re-challenge investigation (figure 6) was showed no significant difference (p
The time course western blot (figure 7) shows a prolonged Erk1/2 activation following exposure of NMU2 to NmU or NmS. The prolonged Erk1/2 activation in the time course western blot could be due to NMU2 internalizing and then recycled back to the surface of the membrane. Internalization is preceded by desensitization. Desensitization occurs to prevent overstimulation of a GPCR.
Generally, this entails the phosphorylation of a receptor. This is conducted by second-messenger mediated protein kinases (such as PKA or PKC) which feedback onto the receptor to reduce second messenger production (Kohout et al., 2003; Irannejad et al., 2014). Alternatively, G-protein coupled receptor kinases (GRK) phosphorylate the receptor.
Phosphorylation by the GRK elicits recruitment of β-arrestin, which is not elicited by second-messenger mediated protein kinases. The receptor itself activates GRKs to phosphorylate serine and threonine residues in the intracellular domains of GPCRs. GRK2-6 regulates the phosphorylation of GPCRs (Shenoy et al., 2011).
Various different phosphorylation combinations result in the receptor increasing its affinity and results in the subsequent binding of different β-arrestin to the receptor (Reiter et al., 2006). This results in the G-protein uncoupling from the receptor and therefore desensitization (Reiter et al., 2006).
Desensitization following GRK phosphorylation is caused by β-arrestin sterically hindering G-proteins, preventing their association with the GPCR to prevent signaling. Also β-arrestin act as scaffolds for kinases which degrade second messengers in desensitization (Nelson et al., 2007).
Following desensitization one function of β-arrestin is to act as an adaptor protein to the GPCR to recruit membrane activated receptors for internalization. Following internalization, the receptor can be degraded, or re-sensitized and returned to the cell surface.
The signalling of the internalized receptor must be terminated to allow re-sensitization or degradation to occur. The affinity of β-arrestin to the receptor was initially shown to regulate the fate of the receptor following internalization. More recently B-arrestin is shown to be part of the actin–sorting nexin 27 (SNX)–retromer tubule (ASTR) complex. The fate of the endosome is regulated by the ASTR complex by forming to act as a molecular brake to reduce internalized receptor signalling.
The ASTR complex consists of many proteins including β-arrestin, retromer, the post-synaptic density (PDZ)-adapter protein SNX27 and the WASH complex. These work together to package the internalized receptor complex into tubules which are then marked for degradation or recycling. The recycling of the receptor occurs via either ASTR-dependent, ASTR-independent or retrograde transport (Pavlos et al., 2016). However, the presence of the ASTR complex is currently limited to the class B parathyroid hormone receptor (PTHR) which form an early endosome (Chan et al., 2016; McGarvey, et al., 2016).
This early endosome can be degraded, can form a signalosome or result in the recycling of the receptor via ASRT dependent or independent recycling. Therefore, NmU may cause NMU2 to undergo desensitization, internalization and re-sensitization subsequently, returning to the cell surface like the PTHR.
Previous work in our lab has shown, using a pH7.4 KHB wash (APPEDIX X) to remove extracellular free ligand, causes a prolonged Erk1/2 activation following NmS stimulation of NMU2. Conversely, NmU stimulation of NMU2 shows a decreasing Erk1/2 activation over time. Comparison of the pH7.4 KHB wash western blot shows Erk1/2 activation decreases over time whereas the time course western blot Erk1/2 activation is sustained following NmU stimulation. This shows NmU Erk1/2 activation requires ligand to be present to allow the re-sensitized NMU2 to bind the extracellular NmU and continue to show a prolonged Erk1/2 response.
However, the Erk1/2 activation following NmS stimulation of NMU2 does not attenuate with the pH7.4 wash. This supports evidence for NmU causing NMU2 to undergo receptor recycling like PTRH, but questions the role of NmS in receptor recycling. Therefore, NmU and NmS are shown to activate Erk1/2 via different pathways.
The western blot time course also shows following NMU2 stimulation by NmS Erk1/2 activation is slightly higher than Erk1/2 activation caused by NmU.
This could explain the purpose of the heighted potency of NmS to NMU2 compared to NmU because the ligand may stay bound to the receptor for longer than NmU. This also provides further evidence to suggest NmU and NmS activate Erk1/2 differently. NmS Erk1/2 activation may be driven by internalized NMU2 signalling in a G-protein independent manner as it does not require extracellular ligand for prolonged Erk1/2 activation. Receptors including but not limited to PTHR, GLPR and V2R have all shown sustained signalling following internalization (Pavlos et al., 2016).
The pH2 acid wash western blot shows evidence for G-protein independent signalling as stimulation of NMU2 by both NmU and NmS resulted in Erk1/2 activation. This suggests both NmU and NmS can cause NMU2 to internalize because this Erk1/2 activation following the acid wash could only be from internalized receptor as the pH2 acid removed free and receptor bound ligand.
Internalization also known as sequestration, is essential to allow G-protein independent signalling. Internalization also allows a GPCR to become re-sensitized or downregulated following desensitization (Reiter et al., 2006). Internalization can occur by three main mechanisms.
Firstly, clathrin-dependent internalization, requires GRK-mediated phosphorylation and β-arrestin binding. This is shown to occur both in the absence and following agonist-receptor stimulation (Oakley et al., 2002; DeWire et al., 2007).
Subsequently, AP-2 adapter protein binds to the carboxy-terminal tail of β-arrestin (Kern et al., 2009). AP-2 aids the recruitment of clathrin to form invaginations of the membrane known as clathrin coated pits (CCP) containing the GPCR with β-arrestin and AP-2 bound.
This CCP is then removed from the membrane by a GTPase known as dynamin and an endosome is formed. This is known as clathrin-dependent internalization. Over time N ethyl maleimide-sensitive fusion protein (NSF), the small G protein ARF6, and PI4P kinase have been identified as binding partners of B-arrestin which facilitate internalization (Nelson et al., 2007; Shenoy et al., 2011).
Also ubiquitination of β-adrenoceptor 2 by E3 ubiquitin ligase is shown to enhance clathrin binding to β-arrestin and stabilizes the endosome more than a non-ubiqintinated endosome (Shenoy et al., 2007). The receptor may also be internalized without initial GRK phosphorylation. This second mechanism of internalization is involving formation of calveolae, instead of clathrin pits.
Alternatively, internalization can occur via uncoated vesicles. The mechanism by which each receptor is internalized is shown to be receptor regulated. This is evident as the CCR5 receptor internalizes in a β-arrestin -dependent manner whereas neuropeptide Y2 receptor internalizes in a β-arrestin -independent manner (Walther et al., 2013). However, most GPCRs internalize in a β-arrestin -dependent and clathrin-dependent manner. Future investigations of NMU2 using confocal microscopy could confirm if NMU2 is internalized following stimulation by NmU and NmS.
In addition to suggesting NMU2 is internalized, the acid wash western blot cell stimulation shows the internalized NMU2 signals. This showed both NmU and NmS cause NMU2 to undergo internalized receptor signalling. This is because an Erk1/2 activation still occurs even when acid wash removes any ligand bound to the receptor. Hence, the lack of ligand bound to the receptor suggests Erk1/2 response could only come from the internalized receptor. An internalized receptor signals by forming a complex known as a signalosome. This represents an intracellular complex within an endosome, composed of the internalized receptor with β-arrestin acting as a scaffold for AP2 and clathrin.
The signalosome mediates G-protein independent signalling and is regulated by the actions of β-arrestin (Walther et al., 2013). Class B receptors which have a high affinity to β-arrestin 1 and 2 are associated with sustained MAPK activation following internalization. Whereas, class A receptors bind β-arrestin 2 preferentially but transiently, hence are mostly associated with re-sensitization of the receptor. Chimeric class A and class B receptors has shown the carboxyl terminal of the receptors regulate the fate of the receptor as class A receptors lack a Ser-Thr rich region in the carboxyl terminal end.
Chimeric receptors containing the carboxyl terminal of class A β2 adrenergic receptor (β2AR) preferentially undergo recycling of the receptor. Chimeric receptors containing the carboxyl terminal of class B V2 vasopressin receptors fail to show re-sensitization but show prolonged MAPK activation, which class A rectors lack (Zhang et al., 1999; Shenoy et al., 2011). This is because class B receptors bind the hormones or β-arrestin in a two-step mechanism by first binding to the N-terminus then the C-terminus and form a stable early endosome which allows internalized receptor signalling (Pavlos et al., 2016).
Therefore, the composition of the C-terminal of the receptor determines the type of β-arrestin recruited. NmS shows internalized NMU2 G-protein independent signalling. NmU shows contrasting pathways for the activation of Erk1/2. This is shown by comparison of the time course western blot and pH7.4 KHB western blot. They suggest NmU must be present to activate Erk1/2. Hence this suggests a G-protein dependent nature. However, the pH2 wash western blot contradicts the requirement of the ligand to activate Erk1/2, hence suggesting a G-protein independent pathway.
The cannabinoid 1 GPCR also shows G-protein dependent signalling initially, which is later replaced by G-protein independent signalling at later time points (Delgado-Peraza et al., 2016). This principle could explain why NmU shows activation of Erk1/2 via both G-protein dependent and independent pathways.
NmS may show more of a preference to G-protein independent signalling compared to NmU. This is because NmS shows Erk1/2 activation via G-protein independent pathways from the pH2 acid wash western blot. However, NmS does not show any G-protein dependent nature because when free ligand is removed using pH7.4 KHB wash the Erk1/2 response remains sustained. As the signalosome is essentially an endosome, G-protein-independent signalling is spatially distinct from the G-protein dependent signalling of the receptor at the cell surface. Therefore, initially the GPCR can activate G-protein dependent signalling.
This will eventually result in the recruitment of β-arrestin. Then when β-arrestin 2 is bound (class B) this can elicit G-protein independent signalling. Therefore, intracellular downstream signalling by the receptor can occur by G-protein dependent or independent signalling. It has been shown that different ligands can cause activation of one of the above signalling pathways more than another. This is known as bias agonism (Rajagopal et al., 2010; Whalen et al., 2011). An example of this is shown by the serotonin receptor. Serotonin is shown to act on the serotonin 2A receptor to cause a β-arrestin mediated Erk1/2 activation and result in head movement in mice. However, a different ligand (DOI) causes the head movement by activation of Erk1/2 in a β-arrestin independent pathway (Schmid et al., 2008). This ligand specific downstream signaling could explain why stimulation of NMU2 by NmU activates Erk1/2 via G-protein dependent and independent mechanisms and NmS stimulates only G-protein independent activation of Erk1/2.
The 5min pH2 acid wash shows a significantly greater Erk1/2 activation compared to the 1min pH2 acid wash for NmU at the 5min and 10min time points (figure 8). This suggests more G-protein-independent Erk1/2 activation occurs after 5min compared to 1min. This is expected as the G-protein independent signalling is shown to take longer to occur compared to the G-protein dependent downstream signalling (Kim et al., 2005; Luttrell et al., 2010)
G-protein independent signalling shown by NmS involves biding of β-arrestin to receptor. To allow high affinity binding of β-arrestin, phosphorylation of GPCRs is shown to be required by many GPCRs such as β2AR, angiotensin II type 1 and a2-AR etc (see review, maybe see actual papers). The dopamine D1A and calcitonin gene-related peptide receptors are shown to associate with GRK5 and GRK6. GRKs can be tissue specific by recruiting multiple different kinases to potentially phosphorylate the receptor at a variety of different combinations of amino acids (Tobin et al., 2008). Also GRK6 is shown to regulate the rate in which the receptor is phosphorylated, specifically in β2AR (Violin et al., 2006). Therefore, each combination of protein kinases or GRKs recruited results in a unique phosphorylation signature and hence as specific signaling outcome (Tobin et al., 2008).
Some GPCRs can also bind β-arrestin independent of phosphorylation, such as D6 chemokinse receptor (Walther et al., 2013). Different β-arrestin bind to different GPCRs with different affinities. The type of GRK involved in phosphorylation and the subsequent β-arrestin recruitment causes different intracellular signals. For example, phosphorylation of the angiotensin II type 1A receptor by GRK5 causes β-arrestin dependent activation of Erk1/2 signaling. However, GRK2 causes β-arrestin dependent endocytosis in the same receptor. (Walther et al., 2013). Therefore, the β-arrestin recruited by the receptor can mediate a specific signalling pathway.
Further investigations could be conducted to isolate the type of β-arrestin recruited for internalization of NMU2 by NmU or NmS. This will help identify the roles of G-protein dependent and independent activation of Erk1/2, by NmU and NmS.
Overall NmU binding NMU2 shows both G-protein dependent and G-protein independent activation of Erk1/2, which raises doubt around the possible recycling of NMU2 in the time course western blot. Alternatively, a more probable theory is NmU shows the ability to selectively activate Erk1/2 by following either a G-protein dependent or independent pathway. NmU shows more G-protein independent activation of Erk1/2 after longer stimulation NMU2 by NmU. Therefore, the duration of NmU exposure to NMU2 may be involved in the selectivity process. NmS may have a slightly higher potency to NMU2 due to a greater activation of Erk1/2. Also NmS binding NMU2 shows a G-protein independent activation of Erk1/2.
Subsequently, by identifying the β-arrestin involved in mediating which intracellular signalling pathway NmU and NmS follow, will enhance the potential of NMU2 to be targeted to treat disorders such as obesity.
References
Austin, C., Lo, G., Nandha, K.A., Meleagros, L. and Bloom, S.R. (1995) ‘Cloning and characterization of the cDNA encoding the human neuromedin U (NmU) precursor: NmU expression in the human gastrointestinal tract’, Journal of Molecular Endocrinology, 14(2), pp. 157-169.
Benzon, C.R., Johnson, S.B., McCue, D.L., Li, D., Green, T.A. and Hommel, J.D. (2014) ‘Neuromedin U receptor 2 knockdown in the paraventricular nucleus modifies behavioral responses to obesogenic high-fat food and leads to increased body weight’, Neuroscience, 258, pp. 270-279.
Brighton, P.J., Szekeres, P.G. and Willars, G.B. (2004) ‘Neuromedin U and its receptors: structure, function, and physiological roles’, Pharmacological reviews, 56(2), pp. 231-248.
Chan, A.S., Clairfeuille, T., Landao-Bassonga, E., Kinna, G., Ng, P.Y., Loo, L.S., Cheng, T.S., Zheng, M., Hong, W., Teasdale, R.D., Collins, B.M. and Pavlos, N.J. (2016) ‘Sorting nexin 27 couples PTHR trafficking to retromer for signal regulation in osteoblasts during bone growth’, Molecular biology of the cell, 27(8), pp. 1367-1382.
Dass, N., Bassil, A., North‐Laidler, V., Morrow, R., Aziz, E., Tuladhar, B.R. and Sanger, G. (2007) ‘Neuromedin U can exert colon‐specific, enteric nerve‐mediated prokinetic activity, via a pathway involving NMU1 receptor activation’, British journal of pharmacology, 150(4), pp. 502-508.
Delgado-Peraza, F., Ahn, K.H., Nogueras-Ortiz, C., Mungrue, I.N., Mackie, K., Kendall, D.A. and Yudowski, G.A. (2016) ‘Mechanisms of Biased beta-Arrestin-Mediated Signaling
Downstream from the Cannabinoid 1 Receptor’, Molecular pharmacology, 89(6), pp. 618-629.
DeWire, S.M., Ahn, S., Lefkowitz, R.J. and Shenoy, S.K. (2007) ‘β-arrestins and cell signaling’, Annu.Rev.Physiol., 69, pp. 483-510.
Fang, L., Zhang, M., Li, C., Dong, S. and Hu, Y. (2006) ‘Chemical genetic analysis reveals the effects of NMU2R on the expression of peptide hormones’, Neuroscience letters, 404(1), pp. 148-153.
Fujii, R., Hosoya, M., Fukusumi, S., Kawamata, Y., Habata, Y., Hinuma, S., Onda, H., Nishimura, O. and Fujino, M. (2000) ‘Identification of neuromedin U as the cognate ligand of the orphan G protein-coupled receptor FM-3’, The Journal of biological chemistry, 275(28), pp. 21068-21074.
Hanada, R., Teranishi, H., Pearson, J.T., Kurokawa, M., Hosoda, H., Fukushima, N., Fukue, Y., Serino, R., Fujihara, H. and Ueta, Y. (2004) ‘Neuromedin U has a novel anorexigenic effect independent of the leptin signaling pathway’, Nature medicine, 10(10), pp. 1067-1073.
Hashimoto, T,. Masui, H., Uchida, Y., Sakura, N. and Okimura, K. (1991) ‘Agonistic and antagonistic activities of neuromedin U-8 analogs substituted with glycine or D-amino acid on contractile activity of chicken crop smooth muscle preparations’, Chemical and pharmaceutical bulletin, 39(9), pp. 2319-2322.
Hedrick, J.A., Morse, K., Shan, L., Qiao, X., Pang, L., Wang, S., Laz, T., Gustafson, E.L., Bayne, M. and Monsma, F.J.,Jr (2000) ‘Identification of a human gastrointestinal tract and immune system receptor for the peptide neuromedin U’, Molecular pharmacology, 58(4), pp. 870-875.
Heiman, M.L., Ahima, R.S., Craft, L.S., Schoner, B., Stephens, T.W. and Flier, J.S. (1997) ‘Leptin Inhibition of the Hypothalamic-Pituitary-Adrenal Axis in Response to Stress 1’, Endocrinology, 138(9), pp. 3859-3863.
Hosoya, M., Moriya, T., Kawamata, Y., Ohkubo, S., Fujii, R., Matsui, H., Shintani, Y., Fukusumi, S., Habata, Y., Hinuma, S., Onda, H., Nishimura, O. and Fujino, M. (2000) ‘Identification and functional characterization of a novel subtype of neuromedin U receptor’, The Journal of biological chemistry, 275(38), pp. 29528-29532.
Hosoya, M., Moriya, T., Kawamata, Y., Ohkubo, S., Fujii, R., Matsui, H., Shintani, Y., Fukusumi, S., Habata, Y., Hinuma, S., Onda, H., Nishimura, O. and Fujino, M. (2000) ‘Identification and functional characterization of a novel subtype of neuromedin U receptor’, The Journal of biological chemistry, 275(38), pp. 29528-29532.
Howard, A.D., Wang, R., Pong, S., Mellin, T.N., Strack, A., Guan, X., Zeng, Z., Williams, D.L., Feighner, S.D. and Nunes, C.N. (2000) ‘Identification of receptors for neuromedin U and its role in feeding’, Nature, 406(6791), pp. 70-74.
Irannejad, R. and von Zastrow, M. (2014) ‘GPCR signaling along the endocytic pathway’, Current opinion in cell biology, 27, pp. 109-116.
Jethwa, P.H., Smith, K.L., Small, C.J., Abbott, C.R., Darch, S.J., Murphy, K.G., Seth, A., Semjonous, N.M., Patel, S.R. and Todd, J.F. (2006) ‘Neuromedin U partially mediates leptin-induced hypothalamo-pituitary adrenal (HPA) stimulation and has a physiological role in the regulation of the HPA axis in the rat’, Endocrinology, 147(6), pp. 2886-2892.
Jones, N.A., Morton, M.F., Prendergast, C.E., Powell, G.L., Shankley, N.P. and Hollingsworth, S.J. (2006) ‘Neuromedin U stimulates contraction of human long saphenous vein and gastrointestinal smooth muscle in vitro’, Regulatory peptides, 136(1), pp. 109-116.
Kern, R.C., Kang, D.S. and Benovic, J.L. (2009) ‘Arrestin2/clathrin interaction is regulated by key N- and C-terminal regions in arrestin2’, Biochemistry, 48(30), pp. 7190-7200.
Kim, J., Ahn, S., Ren, X.R., Whalen, E.J., Reiter, E., Wei, H. and Lefkowitz, R.J. (2005) ‘Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling’, Proceedings of the National Academy of Sciences of the United States of America, 102(5), pp. 1442-1447.
Kohout, T.A. and Lefkowitz, R.J. (2003) ‘Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization’, Molecular pharmacology, 63(1), pp. 9-18.
KUROSAWA, K., SAKURA, N. and HASHIMOTO, T. (1996) ‘Structure-activity relationships of neuromedin U. III. Contribution of two phenylalanine residues in dog neuromedin U-8 to the contractile activity’, Chemical and pharmaceutical bulletin, 44(10), pp. 1880-1884.
Lee, W., Liu, S., Shen, J., Jin, Y., Lai, R. and Zhang, Y. (2005) ‘Identification and molecular cloning of a novel neuromedin U analog from the skin secretions of toad Bombina maxima’, Regulatory peptides, 129(1), pp. 43-47.
Luttrell, L.M. and Gesty-Palmer, D. (2010) ‘Beyond desensitization: physiological relevance of arrestin-dependent signaling’, Pharmacological reviews, 62(2), pp. 305-330.
Martinez, V.G. and O’Driscoll, L. (2015) ‘Neuromedin U: a multifunctional neuropeptide with pleiotropic roles’, Clinical chemistry, 61(3), pp. 471-482.
Maruyama, K., Konno, N., Ishiguro, K., Wakasugi, T., Uchiyama, M., Shioda, S. and Matsuda, K. (2008) ‘Isolation and characterisation of four cDNAs encoding neuromedin U (NMU) from the brain and gut of goldfish, and the inhibitory effect of a deduced NMU on food intake and locomotor activity’, Journal of neuroendocrinology, 20(1), pp. 71-78.
McGarvey, J.C., Xiao, K., Bowman, S.L., Mamonova, T., Zhang, Q., Bisello, A., Sneddon, W.B., Ardura, J.A., Jean-Alphonse, F., Vilardaga, J.P., Puthenveedu, M.A. and Friedman, P.A. (2016) ‘Actin-Sorting Nexin 27 (SNX27)-Retromer Complex Mediates Rapid Parathyroid Hormone Receptor Recycling’, The Journal of biological chemistry, 291(21), pp. 10986-11002.
Micewicz, E.D., Bahattab, O.S., Willars, G.B., Waring, A.J., Navab, M., Whitelegge, J.P., McBride, W.H. and Ruchala, P. (2015) ‘Small lipidated anti-obesity compounds derived from neuromedin U’, European journal of medicinal chemistry, 101, pp. 616-626.
Mitchell, J., Maguire, J. and Davenport, A. (2009) ‘Emerging pharmacology and physiology of neuromedin U and the structurally related peptide neuromedin S’, British journal of pharmacology, 158(1), pp. 87-103.
Mitchell, J.D., Maguire, J.J., Kuc, R.E. and Davenport, A.P. (2009) ‘Expression and vasoconstrictor function of anorexigenic peptides neuromedin U-25 and S in the human cardiovascular system’, Cardiovascular research, 81(2), pp. 353-361.
Mori, K., Miyazato, M., Ida, T., Murakami, N., Serino, R., Ueta, Y., Kojima, M. and Kangawa, K. (2005) ‘Identification of neuromedin S and its possible role in the mammalian circadian oscillator system’, The EMBO journal, 24(2), pp. 325-335.
Nandha, K.A., Benito-Orfila, M.A., Smith, D.M. and Bloom, S.R. (1993) ‘Characterization of the rat uterine neuromedin U receptor’, Endocrinology, 133(2), pp. 482-486.
Nelson, C.D., Perry, S.J., Regier, D.S., Prescott, S.M., Topham, M.K. and Lefkowitz,
R.J. (2007) ‘Targeting of diacylglycerol degradation to M1 muscarinic receptors by beta-arrestins’, Science (New York, N.Y.), 315(5812), pp. 663-666.
Oakley, R.H., Hudson, C.C., Cruickshank, R.D., Meyers, D.M., Payne Jr, R.E., Rhem, S.M. and Loomis, C.R. (2002) ‘The cellular distribution of fluorescently labeled arrestins provides a robust, sensitive, and universal assay for screening G protein-coupled receptors’, Assay and drug development technologies, 1(1), pp. 21-30.
Pavlos, N.J. and Friedman, P.A. (2016) ‘GPCR Signaling and Trafficking: The Long and Short of It’, Trends in Endocrinology & Metabolism, .
Peier, A., Kosinski, J., Cox-York, K., Qian, Y., Desai, K., Feng, Y., Trivedi, P., Hastings, N. and Marsh, D.J. (2009) ‘The antiobesity effects of centrally administered neuromedin U and neuromedin S are mediated predominantly by the neuromedin U receptor 2 (NMUR2)’, Endocrinology, 150(7), pp. 3101-3109.
Raddatz, R., Wilson, A.E., Artymyshyn, R., Bonini, J.A., Borowsky, B., Boteju, L.W., Zhou, S., Kouranova, E.V., Nagorny, R., Guevarra, M.S., Dai, M., Lerman, G.S., Vaysse, P.J., Branchek, T.A., Gerald, C., Forray, C. and Adham, N. (2000) ‘Identification and characterization of two neuromedin U receptors differentially expressed in peripheral tissues and the central nervous system’, The Journal of biological chemistry, 275(42), pp. 32452-32459.
Rajagopal, S., Rajagopal, K. and Lefkowitz, R.J. (2010) ‘Teaching old receptors new tricks: biasing seven-transmembrane receptors’, Nature reviews Drug discovery, 9(5), pp. 373-386.
Rhee, M., Nevo, I., Levy, R. and Vogel, Z. (2000) ‘Role of the highly conserved Asp‐Arg‐Tyr motif in signal transduction of the CB2 cannabinoid receptor’, FEBS letters, 466(2-3), pp. 300-304.
Sambrook, J. and Russell, D.W. (2006) ‘SDS-polyacrylamide gel electrophoresis of proteins’, CSH Protoc, 1, pp. 4.
Scheer, A., Costa, T., Fanelli, F., De Benedetti, P.G., Mhaouty-Kodja, S., Abuin, L., Nenniger-Tosato, M. and Cotecchia, S. (2000) ‘Mutational analysis of the highly conserved arginine within the Glu/Asp-Arg-Tyr motif of the alpha(1b)-adrenergic receptor: effects on receptor isomerization and activation’, Molecular pharmacology, 57(2), pp. 219-231.
Schmid, C.L., Raehal, K.M. and Bohn, L.M. (2008) ‘Agonist-directed signaling of the serotonin 2A receptor depends on beta-arrestin-2 interactions in vivo’, Proceedings of the National Academy of Sciences of the United States of America, 105(3), pp. 1079-1084.
Shan, L., Qiao, X., Crona, J.H., Behan, J., Wang, S., Laz, T., Bayne, M., Gustafson, E.L., Monsma, F.J.,Jr and Hedrick, J.A. (2000) ‘Identification of a novel neuromedin U receptor subtype expressed in the central nervous system’, The Journal of biological chemistry, 275(50), pp. 39482-39486.
Shenoy, S.K. and Lefkowitz, R.J. (2011) ‘β-Arrestin-mediated receptor trafficking and signal transduction’, Trends in pharmacological sciences, 32(9), pp. 521-533.
Shenoy, S.K., Barak, L.S., Xiao, K., Ahn, S., Berthouze, M., Shukla, A.K., Luttrell, L.M. and Lefkowitz, R.J. (2007) ‘Ubiquitination of beta-arrestin links seven-transmembrane receptor endocytosis and ERK activation’, The Journal of biological chemistry, 282(40), pp. 29549-29562.
Thompson, E.L., Murphy, K.G., Todd, J.F., Martin, N.M., Small, C.J., Ghatei, M.A. and Bloom, S.R. (2004) ‘Chronic administration of NMU into the paraventricular nucleus stimulates the HPA axis but does not influence food intake or body weight’, Biochemical and biophysical research communications, 323(1), pp. 65-71.
Tobin, A.B., Butcher, A.J. and Kong, K.C. (2008) ‘Location, location, location… site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling’, Trends in pharmacological sciences, 29(8), pp. 413-420.
van Dijk, G., Donahey, J.C., Thiele, T.E., Scheurink, A.J., Steffens, A.B., Wilkinson, C.W., Tenenbaum, R., Campfield, L.A., Burn, P., Seeley, R.J. and Woods, S.C. (1997) ‘Central leptin stimulates corticosterone secretion at the onset of the dark phase’, Diabetes, 46(11), pp. 1911-1914.
Violin, J.D., Ren, X.R. and Lefkowitz, R.J. (2006) ‘G-protein-coupled receptor kinase specificity for beta-arrestin recruitment to the beta2-adrenergic receptor revealed by fluorescence resonance energy transfer’, The Journal of biological chemistry, 281(29), pp. 20577-20588.
Walther, C. and Ferguson, S.S. (2013) ‘Arrestins: role in the desensitization, sequestration, and vesicular trafficking of G protein-coupled receptors’, Progress in molecular biology and translational science, 118, pp. 93-113.
Whalen, E.J., Rajagopal, S. and Lefkowitz, R.J. (2011) ‘Therapeutic potential of β-arrestin and G protein-biased agonists’, Trends in molecular medicine, 17(3), pp. 126-139.
Wren, A., Small, C., Abbott, C., Jethwa, P., Kennedy, A., Murphy, K., Stanley, S., Zollner, A., Ghatei, M. and Bloom, S. (2002) ‘Hypothalamic actions of neuromedin U’, Endocrinology, 143(11), pp. 4227-4234.
Zhang, J., Barak, L.S., Anborgh, P.H., Laporte, S.A., Caron, M.G. and Ferguson, S.S. (1999) ‘Cellular trafficking of G protein-coupled receptor/beta-arrestin endocytic complexes’, The Journal of biological chemistry, 274(16), pp. 10999-11006.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Sciences"
Sciences covers multiple areas of science, including Biology, Chemistry, Physics, and many other disciplines.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: