Diagnosing and Managing Multiple Endocrine Neoplasia
Info: 6281 words (25 pages) Dissertation
Published: 9th Dec 2019
Contents
MEN type 2A, 2B and non-familial MTC
Abstract
BACKGROUND and Aim
Multiple endocrine neoplasia is associated with significant morbidity and mortality. Well-timed recognition of MEN coupled, together with treatment of associated tumours, results in an improved outcome. The NET clinic at Royal Liverpool University Hospital was established with the aim of providing comprehensive, effective, expert, integrated, multidisciplinary care to patients with NET, including those with MEN. This project looks into the current practice of diagnosing and managing MEN patients, and the scope of any recommendations that can be suggested to improve clinical practice.
Method
The study has been conducted to establish the extent of compliance with international guidelines. The projkect involved a retrospective case note review of all electronic patient records. Electronic records were accessed on ICE, PENs and EPRO. The list of patients was extracted from EPRO by applying the search terms “MEN” and “Multiple Endocrine Neoplasia”. The search captured patients under endocrinologists, general surgery and gastroenterologists.
RESULTS
The search resulted in 38 patients, 35 MEN1 and 3 MEN2. Patients had a total of 51 MEN1 associated tumours that had been diagnosed and treated, either currently, previously or both. Of these patients, only 18 had undergone confirmatory genetic testing. We have also noted that there is no consistency in how imaging is scheduled.
CONCLUSION
MEN patients under the care of RLBUHT are receiving good quality care led by field experts. Nearly all patients are seen by a consultant, they are followed up frequently and families are tested whenever it is possible. However, patients are still seen separately by different experts as needed. As relatives undergo screening, the diagnosis of MEN is likely to increase. The ever increasing numbers of patients requiring screening, surveillance and treatment has implications in the planning of future service provision.
Introduction
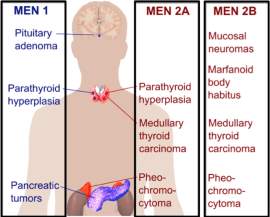 Multiple endocrine neoplasia (MEN) is distinguished by the development of tumours involving two or more endocrine glands within the same patient. The disease has previously been called pluriglandular syndrome. However, glandular hyperplasia and malignancy may also manifest, therefore the name multiple endocrine neoplasia is now
Multiple endocrine neoplasia (MEN) is distinguished by the development of tumours involving two or more endocrine glands within the same patient. The disease has previously been called pluriglandular syndrome. However, glandular hyperplasia and malignancy may also manifest, therefore the name multiple endocrine neoplasia is now
favoured. There are four major recognised types of MEN (MEN1–MEN4), and each type is characterised by the occurence of tumours within particular endocrine glands as seen in the picture above.
 These types of MEN are inherited as autosomal-dominant or they may sporadically manifest; that is, the absence of a family history.1 However, this distinction between familial and sporadic cases is not as straight forward as it seems, because in certain sporadic cases, a full family medical history may be difficult to obtain because the parent with the disease may have passed away before developing any symptoms. MEN1, also known as Wermer’s syndrome2 is characterised by the joint tumours of parathyroid glands, anterior pituitary and pancreatic islet cells. On top of these glands, collagenomas, lipomatous tumours, facial angiofibromas and adrenal cortical tumours have been observed in patients with MEN1.3 However, in MEN2, which is also referred to as Sipple’s syndrome and was previously called MEN2A, medullary thyroid carcinoma (MTC) occurs alongside tumours of parathyroid glands and phaeochromocytoma.2 In MEN3, which was previously known as MEN2B, MTC and phaeochromocytoma occur alongside mucosal neuromas, a marfanoid habitus, dysfunction of the intestinal autonomic ganglion leading to megacolon, and medullated corneal fibres, with uncommon occurrence of parathyroid tumours.2 MTC may also develop as the sole expression of MEN2, and this variant is referred to as MTC-only. In MEN4, also referred to as MENX, patients have anterior pituitary and parathyroid tumours, which may develop in association with tumours of the adrenals, reproductive organs, and kidneys. For the purpose of this report, we will use the terms MEN1, MEN2A and MEN2B.
These types of MEN are inherited as autosomal-dominant or they may sporadically manifest; that is, the absence of a family history.1 However, this distinction between familial and sporadic cases is not as straight forward as it seems, because in certain sporadic cases, a full family medical history may be difficult to obtain because the parent with the disease may have passed away before developing any symptoms. MEN1, also known as Wermer’s syndrome2 is characterised by the joint tumours of parathyroid glands, anterior pituitary and pancreatic islet cells. On top of these glands, collagenomas, lipomatous tumours, facial angiofibromas and adrenal cortical tumours have been observed in patients with MEN1.3 However, in MEN2, which is also referred to as Sipple’s syndrome and was previously called MEN2A, medullary thyroid carcinoma (MTC) occurs alongside tumours of parathyroid glands and phaeochromocytoma.2 In MEN3, which was previously known as MEN2B, MTC and phaeochromocytoma occur alongside mucosal neuromas, a marfanoid habitus, dysfunction of the intestinal autonomic ganglion leading to megacolon, and medullated corneal fibres, with uncommon occurrence of parathyroid tumours.2 MTC may also develop as the sole expression of MEN2, and this variant is referred to as MTC-only. In MEN4, also referred to as MENX, patients have anterior pituitary and parathyroid tumours, which may develop in association with tumours of the adrenals, reproductive organs, and kidneys. For the purpose of this report, we will use the terms MEN1, MEN2A and MEN2B.
MEN1
MEN1 incidence has been determined to be 0.25% from postmortem studies. The prevalence is estimated to be between 0.02 and 0.2 per thousand.4 MEN1 occurs in all ages, with a described age range of 5–81 years, with clinical and biochemical features of the disease having manifested in 80% and 98% of individuals, respectively, by the 5th decade.3 5
Clinical features of MEN1 are linked tumours’ sites and to their secretary products. Parathyroid tumours, causing primary hyperparathyroidism, are the most common feature of MEN1 and occur in around 95% of patients with MEN1367 Hyperparathyroidism (HPT) happens in about 90% of patients, endocrine pancreatic tumours in 60% of patients, and pituitary adenomas in 40% of patients.8 Initial manifestation of MEN1 is HPT. However, HPT may not be noticed until disoderdsr of the pituitary and pancreas prompted medical care. The presence of HPT may also be seen when carrying out screening of family members of those with diagnosed MEN1. MEN1 patients with HPT classically have multiple nodular gland hyperplasia.
Anterior pituitary tumours, consisting of corticotrophinomas, prolactinomas, non-functioning adenomas, and somatotrophinomas, occur in approximately thirty percent (30%) of patients. 6910
Pancreatic islet tumours, also known as pancreatic neuroendocrine tumours (NETs) comprise insulinomas, gastrinomas, vaso-active intestinal polypeptidomas (VIPomas) pancreatic polypeptidomas (PPomas), glucagonomas and non-functioning tumours and these are detected in around 40% of MEN1 patients.
Parathyroid tumours are the initial expression of MEN1 in around 90% of patients, and in the remainder, the initial expression may be a prolactinoma or an insulinoma.35 Combinations of these affected glands and their histologies (i.e. single or multiple adenomas of the parathyroid glands, hyperplasia) have been noted to vary in individuals of the same family and identical twins alike.11 MEN1 is an autosomal-dominant disorder in such families, but a sporadic form (no family history) 3 may occur in 8–14% of MEN1 patients.
Establishing a diagnosis of MEN1 is determined by one of the following three criteria12:

In general, managing MEN1 patients is the same as for each sporadic tumour involved in the syndrome (partathyroidectomy, bromocriptine therapy, other types of surgeries involving the pancreas and the pituitary…). Therefore, the surgical treatment is very much reliant on the phenotypic manifestation in the individual patient. MEN1 patients must have a lifelong follow-up of the adrenal glands, endocrine pancreas, bronchial carcinoids, parathyroid glands, thymus, and the pituitary gland. Because the many components may present in sequence, surgeries of different endocrine organs may be needed over several years.4
MEN1 associated mortality studies have shown that 28–46% of deaths are directly related to MEN1, mainly as a consequence of invasive gastrinomas, foregut carcinoids and pancreatic islet tumours.13 Furthermore, these losses happen at considerably earlier ages than those occurring in non-affected individuals. 13Additionally, a multicentre research from France and Belgium has noted that around 70% of MEN1 patients presently die of MEN1 related causes.14 Thymic carcinoid tumours and malignant pancreatic islet tumours were associated with a marked increased risk of death.14 15
MEN type 2A, 2B and non-familial MTC
MEN2 syndromes are autosomal dominant disorders. Therefore, offspring of affected individuals have a 50% chance of getting the abnormal gene. Biochemical screening has shown nearly 100% penetrance, but only 60-70% develops clinically apparent features.16 Around a quarter of patients with MTC express one of these familial syndromes. Because of rarity of MTC, the incidence of two MTCs in the same family must draw the suspicions to a hereditary syndrome. Approximately 23% of phaeochromocytoma patients also have a hereditary disorder, mainly either von-Hippel-Lindau disease or MEN2.1718
Biochemically, there is no difference between tumours that arise in patients with these genetic patterns and those with the sporadic manifestations of the tumours. Biochemical markers that allow making this distinction is non-existent.
Familial MTC tends to occur at a younger age than sporadic MTC. The clinical manifestation and behaviour vary with each of the features. MTC’s peak incidence is in the 2nd and 3rd decades for MEN2A patients. The tumour commonly progresses slowly, even though it can show early spread. The survival rate for patients with MEN2A and MTC is slightly better than for those with sporadic MTC. A small subcategory of patients with MEN2A has an aggressive form of MTC. Consequently, an aggressive surgical intervention early on is needed to avoid extensive metastases.
MTC in MEN2B occurs in the 1st and 2nd decades of life and inclines to behave more aggressive and fatal19; metastatic diseases leading to death has been observed in young children.20 As soon as MEN2B is diagnosed, surgical treatment for MTC should be performed, with no delay, even in infants. .
In non-MEN familial MTC, the peak incidence is in the 4th to 5th decades. MTC in this condition tends to behave in the least aggressive way.21 Although metastases of the lymph nodes are common, the disease usually follows a slow course, and virtually never causes death.
Phaeochromocytoma in MEN2A and MEN2B has a peak incidence in the 4th and 5th decades. While it can be identified at the same time as MTC, phaeochromocytoma usually develop at a later age than the thyroids.22 Sometimes, phaeochromocytoma is detected in infancy period. Often, these tumours are bilateral and multiple. They are rarely malignant.23 Typically, noradrenaline to adrenaline ratio is lower in the hereditary forms of phaeochromocytoma than in the sporadic form. Symptoms in early stages commonly include headaches, palpitations and anxiety. Screening family allows identifying patients with no symptoms from the tumour. In patients in whom the syndrome is unrecognised, inducing a hypertensive crisis an operation or by childbirth is a risk.
Primary HPT in MEN2A is usually diagnosed at around the age of 37 years; most patients (68%) are found to be asymptomatic at time of diagnosing.24
Patients diagnosed with either MTC or phaeochromocytoma should be screened for the risk of MEN2. Since about 40% of MEN2A gene carriers do not have disease manifestation, a negative family history is not enough to exclude MEN2A. Therefore, members of the family should undergo screening for both tumours. In one research, 174 patients with MTC were screened for phaeochromocytoma, 5 cases were identified in known MEN2 families, and another 5 index cases were found, leading to the discovery of 5 new MEN2 families.25
Biochemical testing is similar to those with the sporadic form of the syndrome. Patients with MTC have increased serum calcitonin. Phaeochromocytoma is known by raised urinary epinephrine, norepinephrine, or VMA. However, plasma normetanephrine and metanephrine may be more sensitive at identifying phaeochromocytoma than urinary testing.26 Patients with subclinical MTC usually have low serum calcitonin levels. However, patients with subclinical MTC or C-cell hyperplasia have a surge in calcitonin levels in reaction to calcium and/or pentagastrin.
Management of MEN2 include surgery. However, patients should be tested for probable phaeochromocytomas, before having thyroidectomy for MCT. The reason behind it is because surgery for phaeochromocytoma takes priority over any neck-related surgery, to prevent a possible dangerous hypertensive crisis during anaesthesia.
Bilateral disease of the adrenals should be treated with bilateral adrenalectomy. Patients with one affected adrenal gland should have unilateral adrenalectomy. Cortical-sparing adrenalectomy is also a possibility in patients who need bilateral adrenalectomy, leading to corticosteroid independent patients in 65% of cases, and a low recurrence rate (around 10%).27
The familial form of MTC necessitates a total thyroidectomy. This will allow to prevent any clinical signs of the disease from developing, therefore increasing the cure rate.
Background
The NET clinic at the Royal Liverpool and Broadgreen University Hospital (RLBUH) was created with the objective of offering complete, effective, united, multidisciplinary care to patients with NET, including those with MEN. The team comprises endocrinologists, gastroenterology specialist, an endocrine surgeon, hepatobiliary surgery, nuclear medicine and radiology. There is also collaboration and input from clinical genetics. The service is consultant delivered.
However, with the increasing number of MEN patients, improvement in genetic testing and the complexity of MEN cases, the need to scrutinise the management of these patients has taken a greater importance. There is a greater importance to draw from other centres’ experiences to deliver high standard of care.
A successful experience of establishing a MEN clinic was observed in Aintree hospital. In accordance with international practice, a multidisciplinary clinic was formed in 2002, with the intent of offering a specialised multidisciplinary service to MEN patients from the areas of Merseyside, Cheshire and North Wales.28
The team at the Aintree clinic consists of two endocrinologists, a paediatric endocrinologist, an endocrine surgeon, and a clinical geneticist. Regular input from hepatobiliary surgery, gastroenterology, radiology and nuclear medicine is also sought. Service provision is consultant-led and interestingly all clinicians are present together in the same room during each consultation. This allows a establishing an individual’s comprehensive management plan with each patient, leading to an integrated approach, and thus avoiding any miscommunication with the patient and between specialities that may otherwise arise as a result of multiple individual discussions. Furthermore, a detailed genetic counselling is provided by the genetics doctor and arrange for family tracing. Typically a further meeting between the patient, family members and the geneticist is required.28
Since early descriptions and explanations of MEN and identification of genetic transformations, awareness of the variable clinical manifestation of MEN was in the increase. This is compounded with a large amount of published case reports. The Journal of Clinical Endocrinology and Metabolism (2001) published the first consensus statement concerning management of patients with MEN and emphasised the significance of a systematic, evidence-based approach. The proposed guidelines by the MEN consensus statement were designed to improve diagnosis and management of MEN patients.7 There are no NICE guidelines regarding the management of MEN.
Aim
The aim of this project is to see whether the current practice of diagnosing and managing patients with MEN who attended the royal Liverpool Broadgreen University Hospital Trust is in line with current recommendations from the literature available and the experience in Aintree Teaching Hospital.
The findings will be critically evaluated and recommendations will be set out.
Methods
This project is a retrospective research. The list of patients was extracted from EPRO database by applying the search terms “MEN” and “Multiple Endocrine Neoplasia”. The search captured all patients under endocrinologists, general surgery and gastroenterologists; 35 patients with MEN1 and 3 patients with MEN2.
ICE was used to view blood test and scan results. PENS, which is the electronic patient notes was also used to view any conversation that may have taken place in clinic.
Excel was used to input data collected which included information such:
| Data Category | Value |
| Date of Birth (DOB) | DD/MM/YYYY |
| Tumour in principal MEN1 related organ | Parathyroid, pancreas, pituitary |
| Imaging | MRI, CT scan, Dual phase, Gallium |
| Genetic Testing | Y/N/Unknown |
| Family Tracing | Y/N/ Unknown |
| Medical Treatment | Somatostatin analogue, bromocriptine… |
| Surgical Treatment | Parathyroidectomy, thyroidectomy, pancreatectomy… |
| Follow up | Blood, Imaging, gut hormones (frequence in months 3,6…). S |
| Seen by | Consultant, registrar, ST1-7, Physician Assistance, Specialist Nurse |
Results
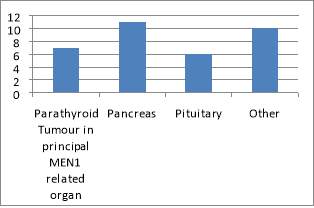 The ages of patients vary between 17 and 77. The mean age of the population is 48.7 and the median is 51.7.
The ages of patients vary between 17 and 77. The mean age of the population is 48.7 and the median is 51.7.
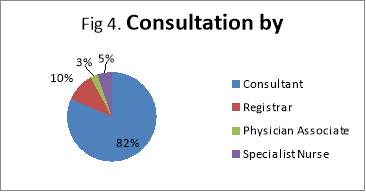
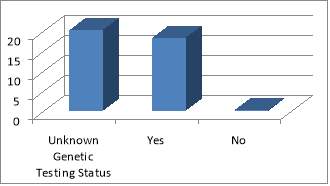 The main organ affected by MEN1 is the pancreas followed by NET of the upper GI organs (small bowels and appendix). The population ages vary from 35 MEN1 patients under the care of NET clinic at the Royal had a total of 51 MEN1 associated tumours that had been diagnosed and treated, either currently, previously or both. The endocrinopathies includes 11 of hyperparathyroidism, 11 pituitary tumours, 26 pancreatic/upper GI neuroendocrine tumours and 3 adrenal adenomas. Of the 38 patients studied, there were 35 confirmed cases of MEN1. Of these patients, only 18 had undergone confirmatory genetic testing; the rest of patients have an unknown genetic status. Equally it is not clear if this has taken place during subsequent follow-ups. This does not mean that genetic testing did not take place; it is highly likely that these have taken place somewhere else with no linked system to the Royal hospital, or simply because of inadequate records keeping. Patients were seen primarily by a consultant during their clinic follow-up (82%). All family members identified through genetic testing were offered prophylactic treatment, surgery or monitoring.
The main organ affected by MEN1 is the pancreas followed by NET of the upper GI organs (small bowels and appendix). The population ages vary from 35 MEN1 patients under the care of NET clinic at the Royal had a total of 51 MEN1 associated tumours that had been diagnosed and treated, either currently, previously or both. The endocrinopathies includes 11 of hyperparathyroidism, 11 pituitary tumours, 26 pancreatic/upper GI neuroendocrine tumours and 3 adrenal adenomas. Of the 38 patients studied, there were 35 confirmed cases of MEN1. Of these patients, only 18 had undergone confirmatory genetic testing; the rest of patients have an unknown genetic status. Equally it is not clear if this has taken place during subsequent follow-ups. This does not mean that genetic testing did not take place; it is highly likely that these have taken place somewhere else with no linked system to the Royal hospital, or simply because of inadequate records keeping. Patients were seen primarily by a consultant during their clinic follow-up (82%). All family members identified through genetic testing were offered prophylactic treatment, surgery or monitoring.
Fig. 2
Fig. 3
Frequency of follow-ups varies considerably. One reason for this is that clinical presentation has changed or patient has developed new symptoms that needed further investigations. Those recorded with “Unknown” under Follow-Up had no records of latest scan or biochemical markers (such as Chromogranin A) or the latest 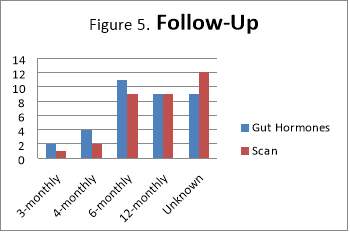 one dates back to at least 2-3 years. However, 76% of patients had repeated gut hormones, including Chromogranin A, as part of the follow up.
one dates back to at least 2-3 years. However, 76% of patients had repeated gut hormones, including Chromogranin A, as part of the follow up.
Two MEN2 patients are from the same family, one of which was successfully traced and offered prophylactic parathyroidectomy and thyroidectomy. All patients, but one, with parathyroid glands as primary site for MEN had calcium levels monitored.
Discussion
The results obtained from the research shows that there is an attempt to centralise care of MEN patients at the royal Liverpool Hospital. Patients are discussed at a regional NET MDT meeting and also seen in the neuroendocrine clinic lead by an endocrinologist as suggested by experts’ guidelines. In the analysis, it was noted that there no consistency in the way patients were followed up or offered biochemical or imaging investigations. For instances, there were cases when patients had CT scan, CT dual phase and MRI all done within 3 months. Given the nature of progression of MEN, it is important that patients are followed consistently and systematically. The guidelines suggest 3-6 months intervals with a yearly biochemical test and once every 2 years for imaging. First degree family members should be reviewed yearly for any signs of disease manifestation.29 Mutation analysis should start as soon MEN is suspected, and there should be no exclusion to any members of the family for genetic testing, including infants. The earliest report of MEN1 manifestation is a pituitary adenoma in a 5-year-old child. Thus, in theory, mutation analysis could be carried out from the age of five. 30
Primary hyperparathyroidism (HPT) is the commonest MEN1 endocrinopathy, attaining nearly 100% penetrance by the age of 50 year.31 However, MEN1 has low prevalence, amounting to only 2–4% of all cases of primary HPT.32 HPT is usually the first clinical expression of MEN1, with a typical age of onset of 20–25 yr.31 Biochemical testing for HPT in MEN1 is central in the recognition of parathyroid tumours (Figure 6). The table below (Figure 6) also shows the different biochemical and imaging testing for each tumour and the appropriate interval.
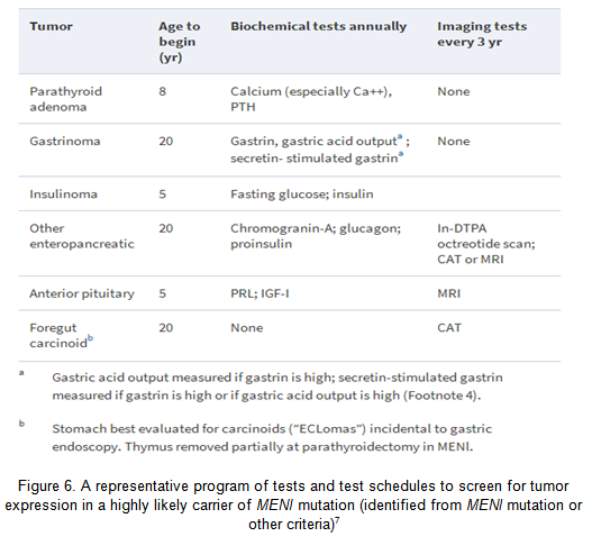
Experts diverge about the timing of parathyroidectomy in MEN1 vs. in sporadic cases. Early parathyroidectomy may reduce the long term exposure to HPT, on one hand; on the other hand, surgery delay may promote a simpler procedure/s. Moreover, despite parathyroidectomy having been done in childhood in MEN1, paediatric indications for surgery have not been ascertained. Imaging of the parathyroid tumour in the preoperative stage has little role in the non-operated MEN1 case due to the need of examining all four glands. Preoperative imaging can be helpful before parathyroid reoperation, and the Tc99m sestamibi scan is the most useful modality.33
Although the number of patients captured during the search was 38 (MEN1 and MEN2), the number is thought to be higher. The reason is because patients’ records are not kept under one central database, but also because they are not under the care of one single clinician (endocrinologist, surgeons, gastroenterologist, oncologist…). In addition, given that both RLBUHT and Aintree hospital look after MEN patients, it is important that improve communication. Ideally, both teams should unite and create a central database for both centres so that each service can access the other service’s patients. The rationale behind it is because families are split between the two centres for many reasons like geographical proximity, convenience, transport, availability of a carer, etc. having access to the database will facilitate access records of family members when needed.
During data collection, it was difficult to identify the main MEN1 organ from the notes. For instance in one patient, the only organ identified was wrongly identified as the Liver. This was a metastatic lesion. Good clinical records provide prospects to improve patient care, integrate performance measures in clinical practice, and enable clinical research. In addition, good records are useful sources of data to help comparative effectiveness studies and new trial designs that may well resolve relevant clinical questions as well as increase efficiency and reduce the cost of endocrinological research.34 In an effort to improve the quality of clinical records, a new template to be used in MEN clinics was devised to be recommended (Appendix 1).
Conclusion
In conclusion, MEN patients under the care of RLBUHT are receiving good quality care led by field experts. Nearly all patients are seen by a consultant, they are followed up frequently and families are tested whenever it is possible. However, patients are still seen separately by different experts as needed. MEN1 patients necessitate multipart care and evidence-based research stipulates that patients are better managed by specialised teams in tertiary centres with crucial members rather than on a case-by-case basis with varying access to facilities and expertise. However, the model is presented with challenges such as the constant increase in numbers of patients needing screening, treatment and surveillance. This has repercussions in the planning of future service provision. Once identified, patients with endocrinopathy must attend the MEN clinic regularly so that surgery can be planned.
At the RLBUTH, a subset clinic of the NET clinic, MEN clinic, should be established to reflect the needs of the patients it serves. The clinic will also have a 3-monthly joint MDT designed to discuss MEN patients. This will be a comprehensive and integrated clinic with the primary aims to evaluate suspected cases, assess family histories, offer genetic counselling and testing, and the creation of a plan of regular endocrine surveillance for MEN patients. Accomplishment of these aims could potentially make an influence on the continuity of clinical management and holistic care of the patients attending the clinic as has been observed in the successful story of the Aintree clinic.28
References
1. Thakker R V. Multiple endocrine neoplasia type 1 (MEN1). Best practice & research Clinical endocrinology & metabolism 2010;24(3):355-70.
2. Thakker R V. Multiple Endocrine Neoplasia—Syndromes of the Twentieth Century. The Journal of Clinical Endocrinology & Metabolism 1998;83(8):2617-20.
3. Trump D, Farren B, Wooding C, et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM : monthly journal of the Association of Physicians 1996;89(9):653-69.
4. Feliberti E, Perry R R, Vinik A. MULTIPLE ENDOCRINE NEOPLASIA TYPE I and MEN II. In: De Groot LJ, Chrousos G, Dungan K, et al., eds. Endotext. South Dartmouth (MA): MDText.com, Inc., 2000.
5. Thakker R V, Bouloux P, Wooding C, et al. Association of parathyroid tumors in multiple endocrine neoplasia type 1 with loss of alleles on chromosome 11. The New England journal of medicine 1989;321(4):218-24.
6. Benson L, Ljunghall S, Akerstrom G, et al. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. The American journal of medicine 1987;82(4):731-7.
7. Brandi M L, Gagel R F, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. The Journal of clinical endocrinology and metabolism 2001;86(12):5658-71.
8. Eberle F, Grun R. Multiple endocrine neoplasia, type I (MEN I). Ergebnisse der inneren Medizin und Kinderheilkunde 1981;46:76-149.
9. Calender A, Giraud S, Lenoir G M, et al. [Hereditary multiple endocrine neoplasia. New genetic data and clinical applications in type 1 multiple endocrine neoplasia]. Presse medicale (Paris, France : 1983) 1995;24(11):542-6.
10. Marx S, Spiegel A M, Skarulis M C, et al. Multiple endocrine neoplasia type 1: clinical and genetic topics. Annals of internal medicine 1998;129(6):484-94.
11. Flanagan D E, Armitage M, Clein G P, et al. Prolactinoma presenting in identical twins with multiple endocrine neoplasia type 1. Clinical endocrinology 1996;45(1):117-20.
12. Turner J J O, Christie P T, Pearce S H S, et al. Diagnostic challenges due to phenocopies: lessons from Multiple Endocrine Neoplasia type1 (MEN1). Human Mutation 2010;31(1):E1089-E101.
13. Dean P G, van Heerden J A, Farley D R, et al. Are patients with multiple endocrine neoplasia type I prone to premature death? World journal of surgery 2000;24(11):1437-41.
14. Goudet P, Murat A, Binquet C, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d’Etude des Tumeurs Endocrines) cohort study among 758 patients. World journal of surgery 2010;34(2):249-55.
15. Thakker R V. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Molecular and Cellular Endocrinology 2014;386(1-2):2-15.
16. Cohen R g, Campos J-M, Salaün C, et al. Preoperative Calcitonin Levels Are Predictive of Tumor Size and Postoperative Calcitonin Normalization in Medullary Thyroid Carcinoma. The Journal of Clinical Endocrinology & Metabolism 2000;85(2):919-19.
17. Neumann H, Berger D P, Sigmund G, et al. Pheochromocytomas, Multiple Endocrine Neoplasia Type 2, and von Hippel-Lindau Disease. New England Journal of Medicine 1993;329(21):1531-38.
18. Raue F, Frank-Raue K. Multiple Endocrine Neoplasia Type 2. eLS: John Wiley & Sons, Ltd, 2001.
19. Schimke R N. Multiple endocrine neoplasia: How many syndromes? American Journal of Medical Genetics 1990;37(3):375-83.
20. Kaufman F R, Roe T F, Isaacs H, Jr., et al. Metastatic medullary thyroid carcinoma in young children with mucosal neuroma syndrome. Pediatrics 1982;70(2):263-7.
21. Farndon J R, Leight G S, Dilley W G, et al. Familial medullary thyroid carcinoma without associated endocrinopathies: a distinct clinical entity. The British journal of surgery 1986;73(4):278-81.
22. Nguyen L, Niccoli-Sire P, Caron P, et al. Pheochromocytoma in multiple endocrine neoplasia type 2: a prospective study. European journal of endocrinology 2001;144(1):37-44.
23. van Heerden J A, Roland C F, Carney J A, et al. Long-term evaluation following resection of apparently benign pheochromocytoma(s)/paraganglioma(s). World journal of surgery 1990;14(3):325-29.
24. Kraimps J L, Denizot A, Carnaille B, et al. Primary hyperparathyroidism in multiple endocrine neoplasia type IIa: retrospective French multicentric study. Groupe d’Etude des Tumeurs a Calcitonine (GETC, French Calcitonin Tumors Study Group), French Association of Endocrine Surgeons. World journal of surgery 1996;20(7):808-12; discussion 12-3.
25. Bonnin F, Schlumberger M, Gardet P, et al. Screening for adrenal medullary disease in patients with medullary thyroid carcinoma. Journal of endocrinological investigation 1994;17(4):253-7.
26. Eisenhofer G, Lenders J W, Linehan W M, et al. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. The New England journal of medicine 1999;340(24):1872-9.
27. Yip L, Lee J E, Shapiro S E, et al. Surgical management of hereditary pheochromocytoma. Journal of the American College of Surgeons 2004;198(4):525-34; discussion 34-5.
28. White H D, Blair J, Pinkney J, et al. Improvement in the care of multiple endocrine neoplasia type 1 through a regional multidisciplinary clinic. QJM : monthly journal of the Association of Physicians 2010;103(5):337-45.
29. Thakker R V, Newey P J, Walls G V, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). The Journal of clinical endocrinology and metabolism 2012;97(9):2990-3011.
30. Stratakis C A, Schussheim D H, Freedman S M, et al. Pituitary macroadenoma in a 5-year-old: an early expression of multiple endocrine neoplasia type 1. The Journal of clinical endocrinology and metabolism 2000;85(12):4776-80.
31. Marx S J, Stratakis C A. Multiple Endocrine Neoplasia – Introduction. Journal of Internal Medicine 2005;257(1):2-5.
32. Uchino S, Noguchi S, Sato M, et al. Screening of the Men1 gene and discovery of germ-line and somatic mutations in apparently sporadic parathyroid tumors. Cancer research 2000;60(19):5553-7.
33. Feo M L D, Colagrande S, Biagini C, et al. Parathyroid Glands: Combination of 99mTc MIBI Scintigraphy and US for Demonstration of Parathyroid Glands and Nodules. Radiology 2000;214(2):393-402.
34. Cowie M R, Blomster J I, Curtis L H, et al. Electronic health records to facilitate clinical research. Clinical Research in Cardiology 2017;106(1):1-9.
Appendices
Appendix 1.

Date
MEN Clinic
Contact details:
PATIENT NAME
ADDRESS
Diagnosis:
Imaging already done:
- At baseline:
- At one year
- At 2 year
Family tracing:
Medication:
Histology of the tumour (if available):
Dear PATIENT
<BODY OF LETTER>
CC.
Patient
Endocrinologist
Geneticist
Surgeon
Gastroenterologist
Oncologist
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Cancer"
Cancer is a disease in which cells grow or reproduce abnormally or uncontrollably. Cancerous cells have the potential to spread to other areas of the body in a process called metastasis.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: