Neuroblastoma Risk Factors and Incidence
Info: 1985 words (8 pages) Dissertation
Published: 16th Dec 2019
Cancer is an abnormal growth of cells in which cells divide and proliferate in an uncontrolled way (Figure 1‑1) caused by multiple mutations and changes in gene expression. These cancerous cells invade the surrounding tissues and can potentially metastasise to distant sites which can lead to death of the host. In a normal condition cells are constantly subjected to signals for controlled cell division, differentiation into another cell or death (due to apoptosis or necrosis). Cancer cells develop resistance to respond to these signals, resulting in uncontrolled growth and proliferation. The characteristic that distinguishes a malignant from a benign tumour is the potential to invade from the site of origin, to enter into systemic circulation, lymph nodes and to metastasize to distant organs in the body (Ruddon 2007).
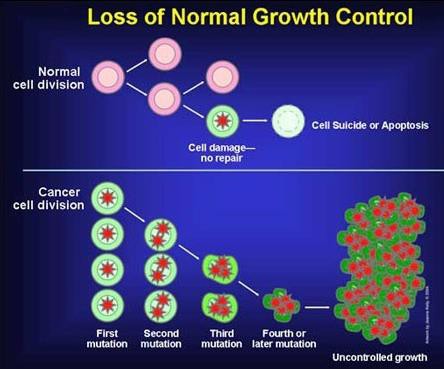
Figure 1‑1 Figure depicting the difference between normal cell and cancer cell division. In cancer cells often the control mechanism that leads to the death of a damaged cell is lost and this leads to accumulation of uncontrolled growth of damaged cell into a tumour (Warburg 1956)
1.1 Hallmarks of cancer
Cancer progression is a multistep process, transforming a normal healthy cell into a highly malignant form via a series of premalignant states into invasive cancers. Hannah and Weinberg et al, described six essential alterations as ‘Hallmark I’ Figure 1‑2 in tumour development. For a normal cell to enter into a neoplastic state, it should acquire the succession of these hallmark capabilities to become tumorigenic and to form malignancy. They include sustained proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis and activating invasion and metastasis (Hanahan and Weinberg 2000).
To clarify and modify the hallmark capabilities in light of progress in cancer research, Hannah and Weinberg et al published new observations with mechanistic concepts and included two enabling characteristics which are important for a cancer cell to acquire the six hallmark capabilities (Hallmark-II). They explained the hallmarks of cancer as acquired functional capabilities that help cancer cells to possess unlimited replicative potential to survive, proliferate and disseminate. In order to acquire these capabilities a cancer cell must possess two emerging characteristics. The first enabling characteristic is the development of genomic instability which leads to random mutations and chromosomal rearrangements. The second prominent characteristic is the involvement of inflammatory state of premalignant and malignant lesions driven by the immune system. This extended concept of hallmarks of cancer helped to understand the complex biology of cancer (Hanahan and Weinberg 2011).
In 2017 Fouad and Aanei published the revisited hallmarks of cancer. Although the hallmarks provide the concept of cancer progression and the alterations for a normal cell to enter into malignant form, it does not answer why these alterations are taking place. As cancer is not a single disease the consequences of disease progression depends on the multiple pathways varied from one tumour type to another. Perhaps these hallmarks of cancer will help in understanding the key for cancer prevention and treatment (Fouad and Aanei 2017). There are approximately 1,800 new cancer cases diagnosed in children in the UK every year. In children, cancer is the most common cause of death accounting 1% of all new cancer cases in the UK. Cancer UK statistics showed around one child in 500 will develop some form of cancer by the age of 14 years.
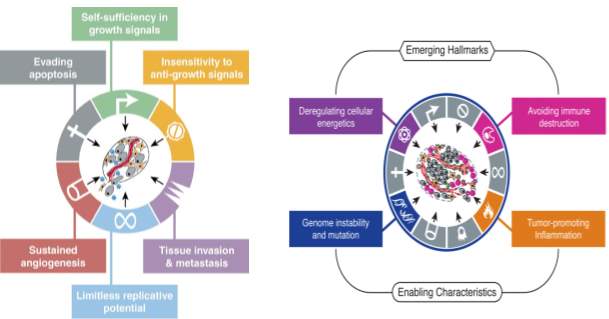
Figure 1‑2 A The six hallmarks of cancer (Hallmarks I) published in 2000 (taken from (Hanahan and Weinberg 2000)) B Emerging Hallmarks and enabling characteristics published in 2011 (taken from (Hanahan and Weinberg 2011))
1.2 Neuroblastoma
1.2.1 Incidence and Age
Neuroblastoma (NB) is a complex, heterogeneous disease (Brodeur 2003) occurring in 10.2 per million children, with 1200 new cases per year diagnosed in Europe and United States. Ninety to 100 cases are diagnosed annually in the UK. The annual incidence is approximately 1 in 105 children less than 15 years of age world-wide. The median age at diagnosis is approximately 22 months, with over one third diagnosed at less than 1 year of age and over 88% diagnosed by the age of 5 (Figure 1‑4). Neuroblastoma originates from the sympatho-adrenal lineage of the neural crest, only present during embryogenesis and this neural crest progresses to form the mature sympathetic nervous system (Stiller, Allen et al. 1995, Brodeur 2003). Spontaneous regression, development of metastasis and responsiveness to treatment depends on the stage of the disease, age of the patient and other molecular, cellular and genetic characteristics of the tumour (Cohn, Pearson et al. 2009). In children under 5 years of age, neuroblastoma usually presents in the abdominal region involving the sympathetic ganglia of the paraspinal region or the adrenal gland (Figure 1‑3). In infants under a year of age, there is a higher incidence of tumours in the thoracic region (Maris 2010). In NB, most of the primary tumours occur within the abdomen (65%). In 1% of patients, the primary tumour cannot be found. Adrenal tumours are more common in children (40%), in comparison to infants (25%). However, there is a higher incidence of thoracic and cervical primary tumours in infants. NB occurs more in boys than girls in the ratio of 1.2:1, this is observed greatest in infant patients (Fisher and Tweddle 2012).
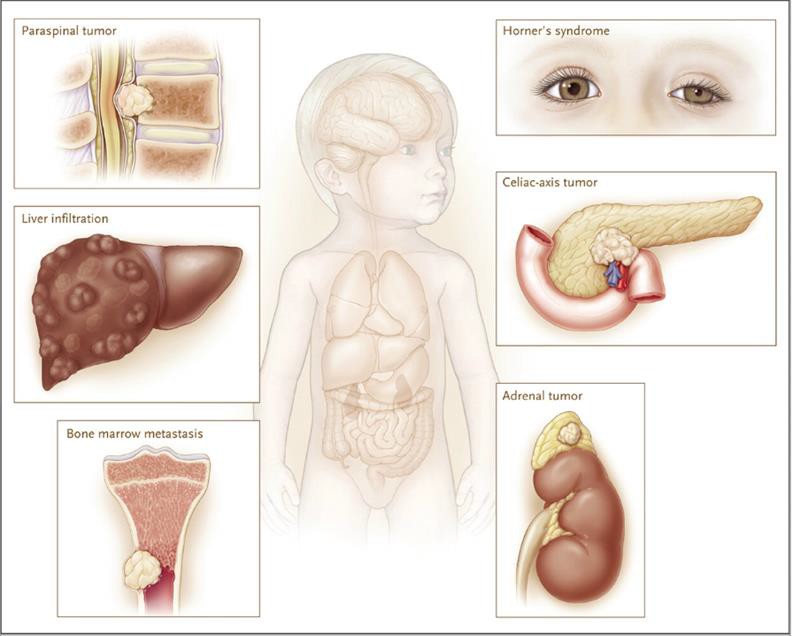
Figure 1‑3 Neuroblastoma tumours originate in most cases in the adrenal gland but can metastasize, affecting other places in the body such as bone marrow, eyes and spinal column (adapted from (Maris 2010))
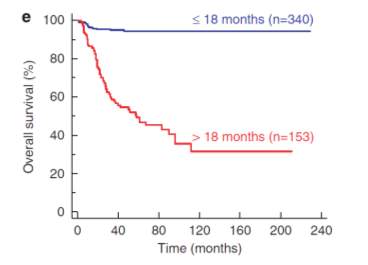
Figure 1‑4 Kaplen-Mierer curve of overall survival of NB patients based on age (Janoueix-Lerosey, Schleiermacher et al. 2010)
1.2.2 Sex
In Neuroblastoma several observations suggest that gender plays is an important factor in determining the natural history of the disease. In a number of pediatric tumours incidence is greater in males than females. The gender and hormonal status may play an important role in the development of neuroblastoma, but a study by Coldman et al assessed the association of sex with survival rate by computing survival curves and confirmed that there was no association between survival and gender (Coldman, Fryer et al. 1980). Based on registries from the United States and Great Britain in children with neuroblastoma, the relative disease incidence in boys when compared with girls was reported to range from 1:1 to 1.4:1 (Hale, Cula et al. 1994).
1.2.3 Aetiology
The aetiology of NB is unknown as it is a heterogeneous disease, a single cause has not yet been identified. However genetic alterations play a more significant role in the development of NB (Fisher and Tweddle 2012).
1.3 Neuroblastic tumours
Neuroblastic tumours arise from the primitive sympathetic ganglion cells. These solid tumours are frequently identified in children (Stiller 2004). They are histologically differentiated based on the number of schwann cells and neuroblasts in the tumour tissue. Depending on the degree of cellular maturation and differentiation, they are categorized as neuroblastoma which is malignant in nature. This is characterized by a diffuse growth of undifferentiated neuroblastic cells separated by irregular thin fibrovascular septa.
Ganglioneuroma a benign tumor in which cells are embedded in a stroma, composed of schwann cells surrounded by neuritic processes and perineurial elements.
Ganglioneuroblastoma (GNB) has intermediate malignant potential according to the International Neuroblastoma Pathology Classification (INPC) system (Shimada, Ambros et al. 1999). Ganglioneuroblastoma can be differentiated as nodular (GNBn) and intermixed (GNBi). A recent report indicated GNB with nodular tumours represents a small proportion of the peripheral neuroblastic tumours. According to INRG (International Neuroblastoma Risk Group) in GNB patients with nodular tumour, the prognosis and treatment response are unfavourable when compared with GNBi patients. This is because most GNBi tumors were localized and non MYCN amplified (De Bernardi, Gambini et al. 2008). Cohn et al reported that patients with GNBn may have very poor prognosis (Cohn, Pearson et al. 2009).
1.3.1 Pathophysiology of NB
To determine the factors which makes a cell susceptible to malignant transformation, it is necessary to understand the link between precursor cells which are responsible for development of disease and molecular, cellular and genetic characteristics of neuroblastoma in later stages. To eradicate cancer and for effective therapy it is essential to target tumour specific mutations. For effective targeted therapy in this heterogeneous disease, it is important to understand the cancer biology between tumour and progenitor cell characteristics.
1.3.2 Neural crest cells: tumor initiating cells
During embryogenesis neural crest cells give rise to diverse cell types such as glial cells, peripheral neuronal cells, Schwann cells, melanocytes and cells of the adrenal medulla. Most neuroblastomas arise from the adrenal medulla and lumbar regions of sympathetic ganglia and the rest arise from chest, pelvic and neck regions (Anderson and Axel 1986, Carnahan, Anderson et al. 1991). Based on the neurochemical and molecular characteristics of neuroblastoma, most of the research into the cells of origin for this heterogeneous disease has concluded that neuroblastoma arises from neural crest cells of the sympatho-adrenal lineage. In the presence of MYCN and bone morphogenetic proteins (BMPs), neuroblast progenitors migrate to the notochord and dorsal aorta from the neural crest and around the neural tube. The cells at this region transform as the primary sympathetic ganglia (PSG) into neural cells of the mature sympathetic ganglia or chromaffin cells (Figure 1‑5).
MYCN is the first mutation observed in a tyrosine hydroxylase (Th)–MYCN-transgenic mouse model, whereas mutations in anaplastic lymphoma kinase (ALK) and paired-like homeobox 2B (PHOX2B) are germline mutations (Trochet, Bourdeaut et al. 2004). A normal sympathetic ganglion cell matures into a terminal ganglion cell or undergoes apoptotic cell death determined by nerve growth factor (NGF), second hit. The pathological state, determined by the postnatal survival of neuroblast precancer cells, requires the cell that is destined to become malignant to be resistant to trophic factor withdrawal before these persistent precancer cells undergo a third change (third hit) to induce transformation, which presents as neuroblastomain early childhood (Marshall, Carter et al. 2014).
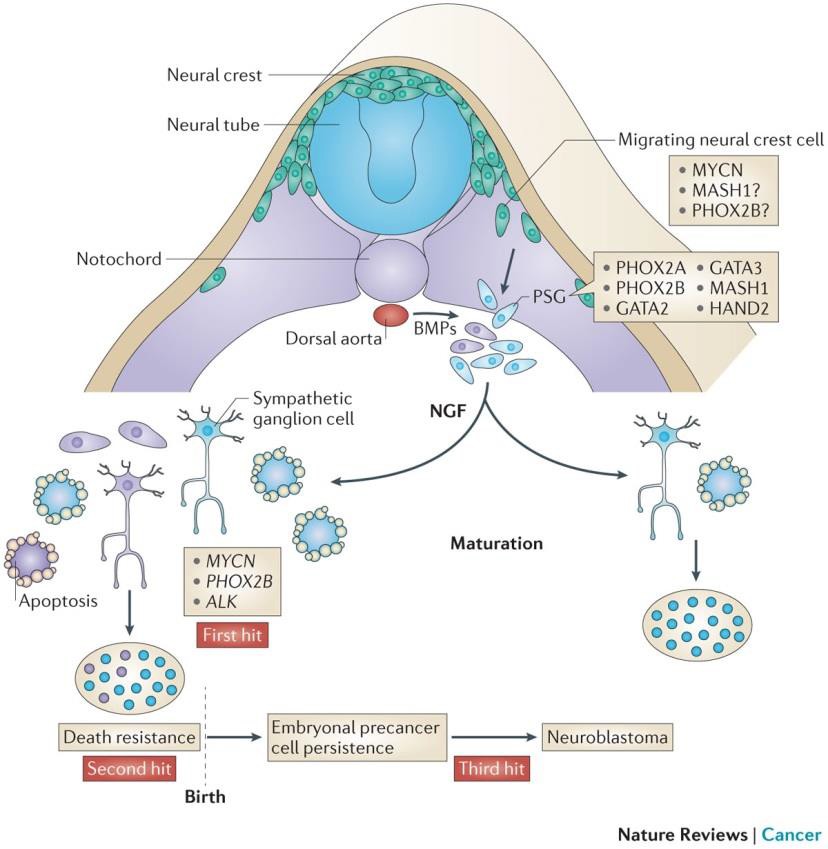
Figure 1‑5 Figure showing the formation of neuroblastoma under the influence of MYCN, PHOX2b and ALk (Marshall, Carter et al. 2014)
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Cancer"
Cancer is a disease in which cells grow or reproduce abnormally or uncontrollably. Cancerous cells have the potential to spread to other areas of the body in a process called metastasis.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: