Why are microbes so much better at making medicines than we are?
Info: 7845 words (31 pages) Dissertation
Published: 21st Feb 2022
Tagged: Biology
ABSTRACT
Over the millennia, Microbes have evolved to synthesise an extensive variety of secondary metabolites to help them survive in harsh environments and to contend with competitor organisms; many such products have proven to be highly useful in combatting human disease. In the last eighty years microbes have emerged as the primary source for the discovery of novel medicines, most notably antibiotics. Although this dominance has waned in recent years, research into the pathways involved in microbial biosynthesis of secondary metabolites (specifically Polyketides and Non-ribosomal peptides) shows the high prolificity and specificity with which these molecules can be produced. The nature of these enzyme systems allows for the synthesis of an array of products so diverse in size and structure it would be impossible for any chemical library produced by humans to match. This research not only indicates the existence of medicines waiting to be discovered, but also provides the possibility of harnessing these pathways to modify existing drugs and even to produce novel chemical scaffolds de novo. An increase in knowledge of these systems, combined with the invention of novel culturing techniques will hopefully facilitate the discovery and production of a new range of medicines.
TABLE OF CONTENTS
List of Abbreviations 1
1 - Introduction 2
1.1 - Microbial Natural products as medicines; an historical overview and a modern perspective 2
1.2 - Microbial Natural Products: an evolutionary advantage 3
1.3 - Classes of Natural Products 4
2 - Medical applications of Natural Products 5
2.1 - Microbial Natural Products as Antibiotics 5
2.2-Other Medical Applications of Microbial Natural Products 5
3 - Polyketides and Non-Ribosomal Peptides as examples of Natural Products 7
3.1 - Polyketides 7
3.2 - Erythromycin A biosynthesis by a Type 1 PKS enzyme 8
3.3 - Non-Ribosomal Peptides 13
3.4 - Biosynthesis of Surfactin by an NRP Synthetase 14
3.5 - Teixobactin, a new NRP 16
3.6 - NRP/PK Hybrid Products 18
3.7 - Targeted modification of Natural Product Synthetases 19
3.8 - NRP/PK Conclusions 20
4 - The way ahead: Discussions and Conclusions 21
4.1 - Natural Products 21
4.2 - Alternatives to Natural Products 22
5 - Summary 24
6 - References 25
LIST OF ABBREVIATIONS
- PK-Polyketide
- PKS-Polyketide Synthase
- 6DEB-6 Deoxyerythronolide B
- DEBS-6 Deoxyerythronolide B Synthase
- NADPH-Nicotinamide adenine dinucleotide phosphate
- LDD-Loading didomain
- AT-Acyltransferase
- ACP-Acyl Carrier Protein
- KS-Keto synthase
- DH-Dehydratase
- ER-Enoylreductase
- KR-Ketoreductase
- TE-Thioesterase
- CoA-Coenzyme A
- NRP-Non-Ribosomal Peptide
- NRPS-Non-Ribosomal Peptide Synthetase
- C-Condensation
- A-Adenylation
- PCP-Peptidyl Carrier
- T-Thiolation
- MT-Methyl Transferase
- TE-Thioesterase
- MRSA-Methicillin Resistant Staphylococcus aureus
- HTS-High Throughput Screening
1 - INTRODUCTION
1.1-Microbial Natural Products as medicines – an historical overview and a modern perspective
Since the advent of medicine, natural products have provided the basis for innumerable treatments. The majority of these are secondary metabolites–molecules synthesised by a limited species range which, although not necessary for life, can convey a survival advantage.[3] The term ‘Natural Product’ encapsulates a diverse range of organic molecules derived from all manner of species. Historically, natural medicines have been predominantly plant or fungal based, however, the advances in microbiology made over the twentieth century have allowed for microorganisms to emerge as a leading source.
While there is evidence to suggest the use of natural products as medicine dating as far back as 60,000 years ago[1], the true golden age of microbial natural product discovery began in 1928 with Alexander Fleming’s discovery of Penicillin. However, there is an increasing worry that this age of discovery may be reaching its limit. Between 1997 and 2007, no new natural products were approved in cancer therapy[5], and despite the increasing number of resistant bacterial strains, for the past 30 years antibiotic discovery has been largely stagnant.[6]
Microorganism-derived natural products have a broad range of pharmaceutical applications as antibiotics, antifungals and cancer treatments. It is only in recent decades, with massive advances in combinatorial chemistry, that alternatives to natural products could be realistically synthesised. Despite these recent advances, natural products still provide the basis for most medicines.[2] In fact structural chemistry is often used simply to produce new drugs through modification of existing natural scaffolds, rather than to synthesise new molecules de novo.[4]
1.2 - Microbial Natural Products: an evolutionary advantage
In general, microbial secondary metabolites can be divided into two subsets–those which provide physiological aide (e.g. by providing resistance to toxins), and those which provide a competitive advantage by hindering the growth of competitor organisms. The rate of production of both types is dependent on the producer’s environment.[9] It is from this second set that the majority of clinically viable antibiotics have been derived, the obvious reason being that these molecules have been evolutionarily optimised to hinder the growth and proliferation of competitor microbes, a characteristic which can be harnessed in medical practices (provided the molecule is not also toxic to humans). The obvious example of this is penicillin, an antibacterial produced by some species of the microscopic fungi of the Penicillium genus which is now used as an antibiotic against a wide variety of bacteria. It is currently difficult for humans to match this level of optimisation using chemical methods.
In the biosynthesis of secondary metabolites it is often the case that all the genes required to produce a single molecule are clustered together in the genome, either as operons or single genes that encode the necessary biosynthetic enzymes. The substrates of these enzymes are often products of primary metabolic pathways. In the case of antibiotics, these clusters often also contain resistance genes that ensure the producer microbe is not affected by the antibiotic biosynthesised. These clusters can account for a significant proportion of the genome; for example, it has been found that the Actinomycete Streptomyces avermitilis contains 30 such clusters which together account for 6% of the total genome.[10]
Given the level of resources and genetic space required to produce many of these secondary metabolites it is clear that their synthesis must convey a significant competitive advantage to the producer, otherwise such complex and metabolically draining pathways could never have evolved.
1.3 - Classes of Natural Products
Natural Products can be classified into 6 main classes:[3]
- Polyketides
- Terpenoids and steroids
- Phenylpropanoids
- Alkaloids
- Non-ribosomal polypeptides
- Specialised carbohydrates
This essay will focus on non-ribosomal peptides and polyketides; their sources, biosynthetic pathways and pharmaceutical applications, with the aim of demonstrating the complexity and precision of the enzymatic pathways that produce modern medicines. I will also compare the different potential methods of developing new medicines; primarily the discovery and harnessing of previously unidentified natural products and processes, the targeted modification of natural molecules, or the development of products produced by combinatorial chemistry.
2 - MEDICAL APPLICATIONS OF NATURAL PRODUCTS
2.1 - Microbial Natural Products as Antibiotics
Since the introduction of Penicillin in the 1940’s, antibiotics have revolutionised the medical industry and saved millions of lives. It is not necessary to detail here the profound impact that the commercial development of antibiotics has had on the treatment of infectious diseases. However, this ‘golden age’ of medicine is under threat; despite the increasing number of antibiotic resistant bacteria very few new antibiotics are being produced. Since the sharp drop off in discovery of new antibiotic classes in the 1970’s an increased emphasis has been put on the chemical modification of existing antibiotic ‘skeletons’ to produce new variations which carry fewer side effects, are more effective and, crucially, are less susceptible to resistance mechanisms.[6] While so far this strategy has proved efficacious, the increasing prevalence of multi-drug resistant bacteria is a significant worry.
There is, however, hope on the horizon, as 2015 saw the discovery of a new antibiotic, Teixobactin. Although still in the early phases of research, teixobactin was shown to have strong bactericidal properties against the bacteria Staphylococcus aureus, Mycobacterium tuberculosis, Clostridium difficile and Bacillus anthracis. This first is especially relevant as the treatment of the common Methicillin Resistant strains of S. aureus currently rely on the use of Vancomycin, an antibiotic with numerous side effects.[14]
The structure and mode of action of Teixobactin will be covered in section 3.2, and its discovery will be covered in section 4.1.
2.2 - Other Medical applications of Microbial Natural Products
Although this essay focuses primarily on the use and production of natural antibiotics, natural products also have many other pharmaceutical applications, most importantly in the treatment of cancer. Approximately three quarters of contemporary anti-tumour drugs were derived from natural products. Similarly to antibiotics, a high proportion of these were isolated from the Actinomycete order of Gram-positive bacteria.[5] Natural products also provide the majority of anti-fungal medicines.
3 - POLYKETIDES AND NON-RIBOSOMAL PEPTIDES AS EXAMPLES OF NATURAL PRODUCTS
3.1 - Polyketides
Polyketides (PKs) are a diverse class of natural products which have a variety of pharmaceutical uses, as antibiotics, antifungals and anticancer drugs. PK’s have a wide structural diversity but can be identified by the presence of the functional groups β-hydroxyketone or β-hydroxyaldehyde. Their representative aromatic chains can range in length from 6 carbons to 168. PK’s are produced in complex biosynthetic pathways by the class of enzyme termed Polyketide Synthases (PKS), usually from the fatty ester primary metabolites such as acetyl-CoA, malonyl-CoA etc. This pathway is analogous to the primary metabolic fatty acid synthesis pathway.[11]
There are three classes of PKS; Type 1 PKS’s, the best studied, are large multidomain structures which contain multiple active sites necessary for the different stages of PK synthesis. Type 2 PKS’s are comprised of separate enzymes which interact to complete synthesis. Type 3 PKS’s are small enzymes found predominantly in plants.
The Tetracycline class of antibiotics provide an excellent example of clinically useful Polyketides. First discovered in 1940 these antibiotics have a broad range of bacterial targets, both Gram positive and Gram negative, and are used to treat a variety of diseases including malaria, syphilis and cholera. They are translational inhibitors which impede protein synthesis, through binding of the 30s ribosomal subunit, thus preventing association of the aminoacyl-tRNA with the A site of the Ribosome. Throughout its decades of use a variety of resistant strains have emerged which work either through drug efflux or ribosomal protection.[12]The tetracycline group of antibiotics all consist of a core of 4 hydrocarbon rings with variable side chains [Fig 1].
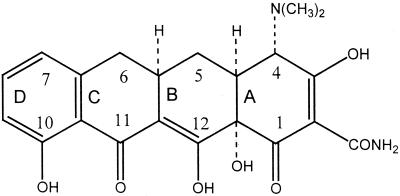
[Fig 1] - The structure of 6-deoxy-6-demethyltetracycline, the most structurally simple tetracycline.
Taken from Chopra & Roberts (2001)[12]
3.2 - Erythomycin A biosynthesis by Type 1 PKS enzymes[18]
Another example of a pharmaceutically useful PK derivative is the versatile antibiotic Erythromycin A (isolated from the bacterium Streptomyces erythreus), which is synthesised from a Polyketide precursor; 6-deoxyerythronolide B (6DEB). Like Tetracycline, Erythromycin A is a translational inhibitor which binds the 50s ribosomal subunit to prevent ribosomal translocation. It is used to treat chlamydia, syphilis and respiratory tract infections.[13] The synthesis of 6DEB by the Type 1 6-deoxyerythronolide B Synthase (DEBS) is a well-studied pathway which will be used here as an example of a Type 1 polyketide biosynthetic pathway[17].
PKS1 enzymes are complex, non-iterative, multidomain enzymes, separated into ‘modules’ each of which is composed of separate domains that provide substrate specificity and function together to perform the elongation steps necessary for PK biosynthesis. DEBS [Fig 2] is a 6-modular enzyme (with an additional loading didomain and Thioesterase domain) which catalyses the polymerization of DEB from one propionyl-CoA and six methyl-malonyl CoA monomers, using 6 NADPH cofactors. Each module contains the domains necessary for one chain elongation step.
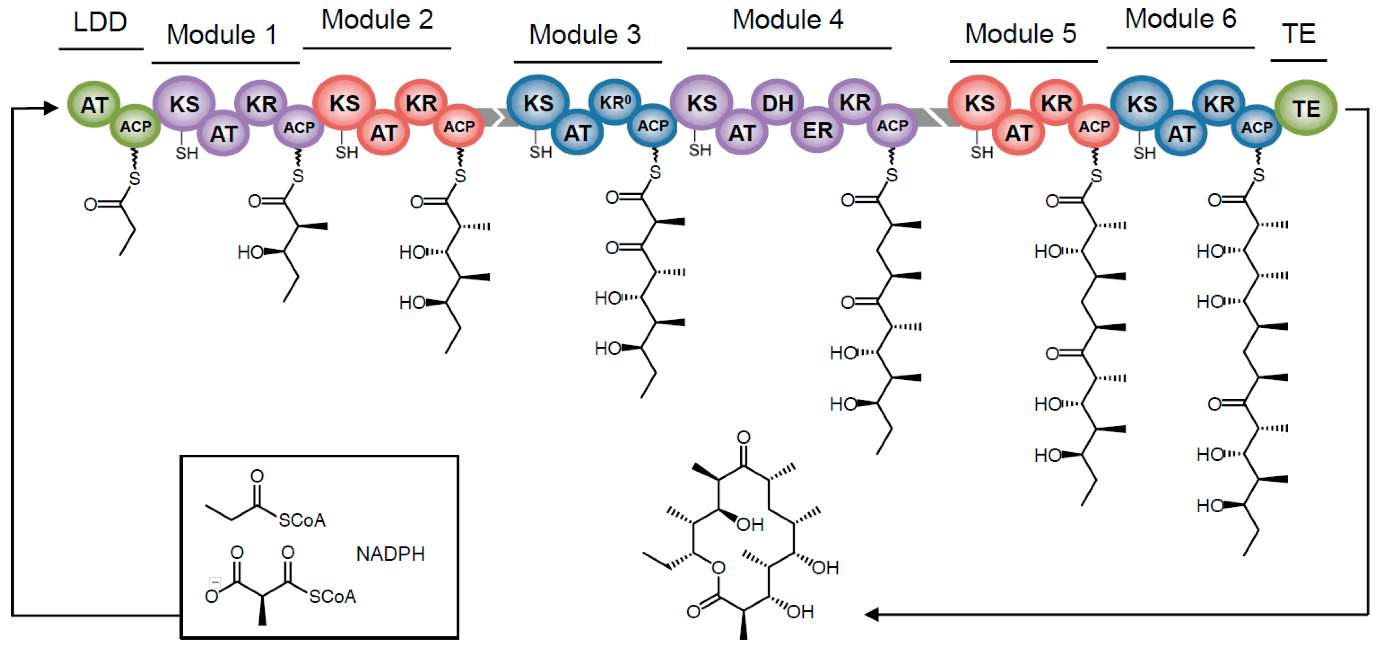
[FIG 2]-Diagram showing the Biosynthesis of 6DEB from 1 propionyl-CoA and 6-methyl-malonyl-CoA by DEBS.
DEBS is comprised of 6 modules with an LDD and a chain terminating TE domain.
KEY:
LDD-Loading didomain AT-Acyltransferase ACP-Acyl Carrier Protein
KS-Keto synthase DH-Dehydratase ER-Enoylreductase
KR-Ketoreductase TE-Thioesterase
Taken from Bayley and Yadav (2017) [29]
Each module of DEBS catalyses the decarboxylation, condensation and reconfiguration of methyl-malonyl-ACP to generate the 2-methyl 3-ketoacyl-ACP intermediate necessary for chain elongation. Depending on the module composition, this intermediate can then be reduced and/or epimerised (switching of one asymmetric centre in a compound to form an epimer, a diastereoisomer with opposite configuration in one stereogenic centre). Both reduction and epimerisation are catalysed by the Ketoreductase domains of the module. In DEBS the combination of these actions are:
- Module 1: Reduction+Epimerisation
- Module 2,5,6: Reduction
- Module 3: Epimerisation
As can be seen in Fig 2 each module must contain a Ketosynthase, Acyltransferase and Acyl Carrier Protein, as these are necessary for function. The ACP recruits the substrate–in this case methyl-malonyl CoA–and therefore also dictates the module’s substrate specificity. The KS domain receives the product from the upstream module. And the AT domain mediates the acyl transfer of the polyketide chain to the substrate. Each ACP must interact with one KS and one AT domain. This KS domain is capable of adopting three distinct conformations;
- KS*: In this conformation the KS domain has a high specificity for the upstream ACP domain. This state is assumed when the ACP and KR domains of the module are both empty.
- KS: No affinity for either ACP- in this state the ACP domain can be loaded with the methyl-malonyl CoA, and the KR can act on this unit.
- KS**: In this state the KS has high affinity for the ACP of the same module, and catalyses chain elongation.
This conformational disparity is necessary for the polyketide chain to be shuttled between modules, and for chain elongation to occur.
The first step in 6DEB biosynthesis is the recruitment of the starter molecule, propionyl CoA by the loading didomain. The LDD is comprised only of an ACP, specific for propionyl CoA, and an AT domain which can catalyse the transfer of this substrate to the appropriate ACP. The subsequent chain is then shuttled through each of the modules, growing as it proceeds. The chain-terminating Thioesterase domain is necessary for release of the final product. The mechanism of action of modules 3 and 4 will be described as examples of module action mechanisms and will be used to show the effect that module composition can have on action. DEBS module 3 [Fig 3] is comprised of the usual KS, ACP and AT domains, with an additional, reductively inactive Ketoreductase domain (KR0).
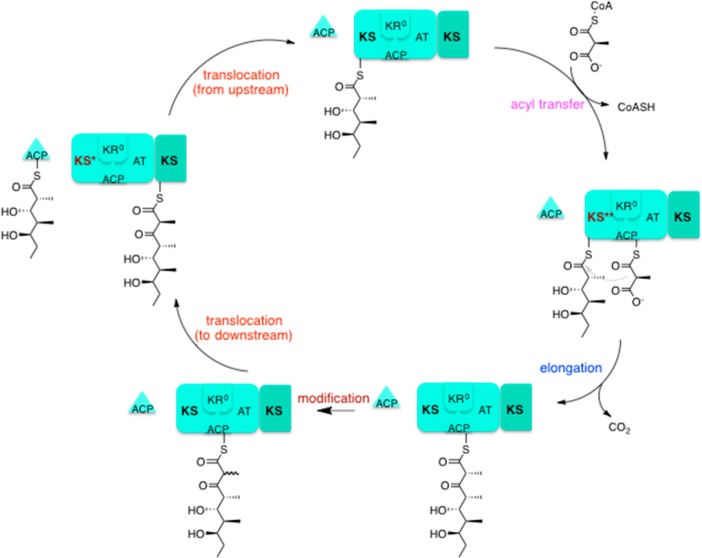
[FIG 3]-Diagram showing the mode of action of 6DEBS Module 3
Taken from Khosla et al (2014)[17]
Chain elongation by module 3 occurs in 5 steps;
- Translocation: The polyketide chain is transferred from the ACP domain of the upstream module 2 to the KS domain of module 3. This process involves transthioesterification
- Acyl Transfer: The AT domain catalyses the transfer of a methyl-malonyl-CoA to the ACP domain.
- Decarboxylation elongation: The polyketide chain is transferred to the methyl-malonyl-ACP. CO2 is released as a by-product.
- Modification: KR0 catalyses epimerisation.
- Translocation: The polyketide chain is translocated to the KS domain of Module 4.
Module 3 is the only domain in DEBS which does not catalyse a reduction reaction – this is due to its inactive KR0 domain. Active KR domains are capable of catalysing 2-methyl epimerization and/or ketoreduction of the substrate. However, KR0 has lost its reductive capacity, and retains only its intrinsic epimerization activity. The lack of Ketoreductase activity results in the inclusion of an additional ketone group.
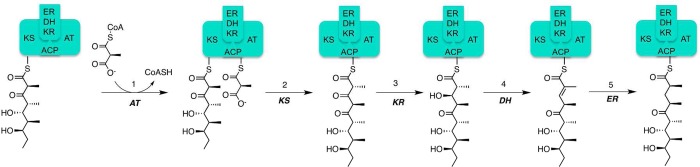
[FIG 4]-Diagram showing the mechanism of 6DEBS Module 4
Taken from Khosla et al (2014)[17]
Module 4 [Fig4] is the most complex module of the DEBS enzyme; in addition to the KS, ACP, AT, and KR domains, it also contains an Enoylreductase (ER) and a Dehydratase (DH) domain. As such, elongation by module 4 contains additional stages, however, the mechanism of transfer of the polyketide chain between modules remains the same.
Stages in chain elongation:
- Translocation
- Acyl Transfer
- Decarboxylation elongation
- Ketoreduction: KR domain reduces a ketone group to form a β Hydroxy intermediate
- Dehydration: DH domain dehydrates this intermediate to form a trans-trisubstituted double bond
- Enoylreduction: The ER domain performs the final reduction of this bond.
- Translocation: polyketide chain transferred to module 5.
The final product of the DEBS Type 1 enzyme has 10 stereogenic centres. Despite there being over a 1000 possible diastereoisomers of this molecule, it is produced with a high level of specificity. This specificity is, of course, genetically encoded and therefore evolutionarily optimised. Structural specificity of the Polyketide product is determined by both the number of modules and their domain composition. The ACP isoform dictates the monomer by which the product will be synthesised, and therefore, along with the number of modules, plays a large role in deciding the final length of the PK. The inclusion of other domains (KR, DH, ER etc) determines the location, composition and orientation of the stereogenic centres. This high level of specificity is unmatched by the majority of industrial total synthesis pathways. Indeed, Erythromycin A is still industrially produced by fermentation of the bacteria Saccharopolyspora erythraea, rather than by semi or total synthesis.[18]
3.3 - Non-Ribosomal Peptides
As their name would suggest, Non-Ribosomal Peptides (NRP’s) are secondary metabolites composed of amino acids synthesised in a ribosomal-independent manner. NRP’s are instead synthesised by large, multimodular enzymes called Non-Ribosomal Peptide Synthetases (NRPS), which mediate sequential condensation reactions necessary to form peptide bonds. These synthetases are structurally and functionally similar to Type 1 Polyketide Synthases. Both enzymes are multimodular, with their specificity and chemical action being dictated by the domain composition of each module. NRP’s are synthesised in both bacteria and fungi. In addition to the 20 amino acids acted on by the ribosome, NRP’s can be synthesised using over 500 different monomers, including α-hydroxy acids, fatty acids and non-proteinogenic amino acids. This monomer variance results in a large structural diversity, which no doubt contributes to NRP function and would explain the need for a non-ribosomal biosynthesis pathway. NRP’s provide a wide variety of pharmaceutically useful compounds including antibiotics (penicillins, cephalosporins), antifungals (bacillomycin), antitumor drugs (bleomycin), and immunosuppressants (cyclosporine).[18]
3.4 - Biosynthesis of Surfactin by an NRP Synthetase
Surfactin is an NRP produced naturally by the bacterium Bacillus subtilis, its synthesis by the Surfactin Synthetase enzyme will be used here as an example of NRP biosynthesis by a modular NRPS. Surfactin is a cyclic lipopeptide with an amino acid sequence; L-Glu-L-Leu-D-Leu-L-Val-L-Asp-D-Leu-L-Leu, with an attached hydrophobic fatty acid chain. There are four isomers of Surfactin (A,B,C and D) which differ in the length of this fatty acid chain. [Fig5]. Surfactin is a powerful surfactant (amphiphilic molecule) which is commonly used as an antibiotic against Gram positive bacteria. Surfactin functions as an antimicrobial primarily by forming pores/cation channels in bacterial membranes. Unfortunately, due to its lack of cellular specificity, Surfactin also has haemolytic effects at high concentrations, which limit its use.[22]
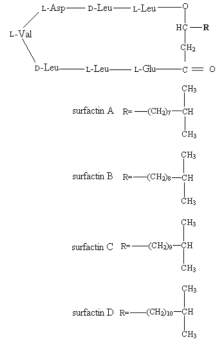
[FIG 5]-Diagram showing the structure of the different Surfactin isoforms
Taken from Shaligram & Singhal (2010) [22]
Surfactin synthetase [Fig 6] is composed of seven modules translated by 3 genes (srfA-A,B&C).
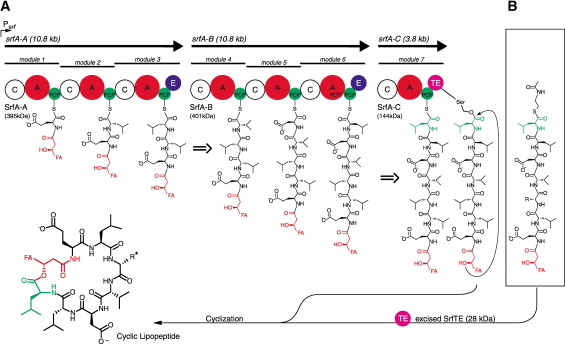
[FIG 6]-Diagram showing the Biosynthesis of Surfactin from its composite amino acids by Surfactin Synthetase.
Taken from Bruner et al (2002)[23]
All central NPRS modules contain a minimum of three domains necessary for polymerisation: a Condensation/C domain, an Adenylation/A domain and a Peptidyl Carrier/PCP domain (alternatively called a Thiolation/T domain). The NPRS initiation module often lacks the C domain, as there is no upstream monomer with which to form a peptide bond. Each of these three domains plays a key role in peptide bond formation. The A domain selects the correct amino acid monomer (its active site composition allows for substrate specificity) and activates it through adenylation. This process uses energy from ATP to produce an amino acyl-adenylate capable of reacting with a thiol on the phosphopantetheinyl arm of the PCP domain to form an activated thioester derivative. The C domain catalyses the condensation reaction between the produced activated monomer with that of the upstream module to form a peptide bond. This assembly line only begins to operate once the A domain of each module has bound its selective amino acid. Once this condition has been met the amino acid of each module is passed sequentially to the upstream module where a peptide bond is formed.
In addition to the obligatory C, A and PCP domains, some modules may also contain an Epimerization (E) domain capable of catalysing the epimerization of amino acids between the L and D enantiomer states. In the case of Surfactin Synthetase, modules 3 and 6 both contain an epimerization domain, which catalyses the epimerisation of L-Leu to D-Leu. Although not found in this particular synthetase, modules in other NRPS enzymes may contain domains with other functions such as a Methyltransferase/MT domain (which catalyses methylation of amide Nitrogen). The C domain may also be switched for a Cy domain which causes the internal cyclisation of Serine, Cysteine or Threonine residues.
The termination module also contains an additional, unique domain, needed to cleave the final product from the enzyme. This is generally a Thioesterase (TE) domain, which breaks the covalent bond between the peptide and the synthetase enzyme by catalysing nucleophilic attack of the bond. In the case of cyclic peptides, such as Surfactin, the nucleophile must be internal. However, in the case of linear peptides the TE domain catalyses nucleophilic attack by a water molecule.[20] Although it was initially believed that the A domain was the only factor involved in module-monomer specificity, it is now believed that the C domain aminoacyl thioester binding site may also have some substrate specificity.
3.5 - Teixobactin, a new NRP
As outlined in section 2.2 Teixobactin is a newly discovered NRP antibiotic which shows great promise in the treatment of infections caused by Gram positive bacteria. It is believed to inhibit the synthesis of the peptidoglycan layer of bacterial cell walls by binding to cell-wall precursors. As peptidoglycan is unique to bacteria, this suggest that it should not cause major harm to eukaryotic cells, indeed laboratory testing showed that it had no toxicity against mammalian cells in vitro. Testing on mice infected with MRSA showed that teixobactin was both an effective antibiotic and safe for use in a mammalian host.[14]
Teixobactin is a cyclic depsipeptide (meaning that it has both peptide and ester linkages) produced by the newly discovered soil bacterium Eleftheria terrae. It is formed through modification of an 11 member peptide chain containing both L and D amino acid stereoisomers, as well as the rare amino acid Enduracididine, which is key for functionality.[15][16] This presents a problem for organic synthesis of Teixobactin; although Enduracididine can be synthesised, the process is difficult and gives a low percentage yield of product.[16]This makes the possibility of synthesizing Teixobactin in a laboratory setting a daunting one.
Teixobactin synthetase [Fig 7] is an 11 module NRPS in which the last 10 modules contain only the core C, A and T (PCP) domains. The first module also contains a methyl transferase (MT) domain which methylates the phenylalanine amide group.
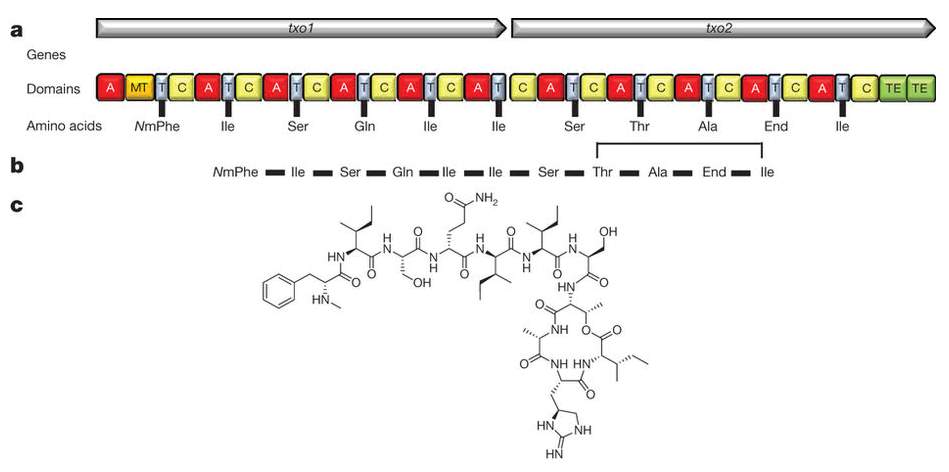
[Fig 7]-Diagram showing the Structure of Teixobactin, its amino acid sequence, and the NRPS which synthesises it.
Taken from Ling et al (2015)[14]
3.6 - NRP/PK Hybrid Products
To further add to the vast array of possible structures that PK/NRP synthetases can produce, there also exists a hybrid class of Natural Product composed of both peptide and fatty acid monomers. These hybrids can be produced by two methods; firstly, the composite polyketide and non-ribosomal peptide can be synthesised separately and fused by a ligase enzyme, e.g. the Pseudomonas syringae phytotoxin, Coronatine. Interestingly, even in the case of biosynthesis by two separate enzymes, the genes necessary to produce this molecule are clustered together, separated only by a regulatory region. Alternatively, these hybrid products can be produced by a ‘hybrid synthetase’ through direct transfer of nascent chains between PKS and NRPS modules in the same enzyme. One example of such a product is the antibiotic Zwittermicin [Fig. 8].[24]
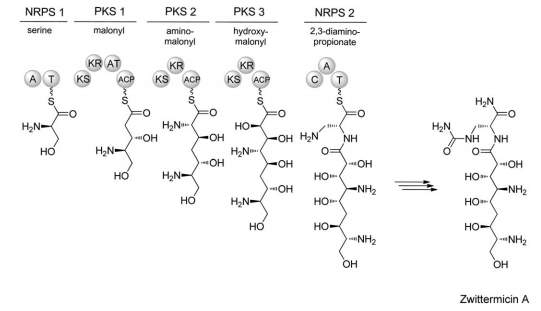
[Fig 8]-A simplified model of the Zwittermicin hybrid synthetase.
Taken from Letzel et al (2013)[25]
3.7 - Targeted modifications of Natural Product Synthetases
Intense study of known NRPS and PKS enzymes has shown a link between substrate selectivity and the active site amino acid sequence of the substrate-selecting NRPS Adenylation domain and the PKS Acyltransferase domains. This knowledge has allowed for the creation of bioinformatics programs (such as antiSMASH) which can identify Natural Product-coding gene clusters through whole-genome scanning, and can then predict NRP/PK monomer sequence, and the resulting product structure, based entirely on the DNA sequence of the identified biosynthesis genes. Although there have proved to be some exceptions to the identified rules, these bioinformatic programs are still highly useful.[25]
This detailed knowledge of the structure and specificity of both PKS and NPRS assembly has allowed for the targeted modification of novel polyketides and non-ribosomal peptides, through module/domain substitution, addition, deletion and modification. In the future this knowledge could allow for the design and biosynthesis of completely novel ‘natural products’. Although no PK/NPRS has yet been synthesised from a completely novel design, domain modification of existing modular synthetases has already proven to be useful. One example of this is the Actinomycete natural product Avermectin, widely used as an insecticide, and as a veterinary/medical drug. Due to its widespread use, resistance to Avermectin has become a serious problem. To overcome this resistance, the synthesis of new analogues was necessary. One research team achieved this by using genetic engineering to switch the substrate specific PKS AT domain in a target module. This caused the switching of one of the PK monomers, resulting in a structurally distinct analogue.[26]
Despite the similarity of the biosynthesis mechanisms, modification of NRP synthetases has proved less fruitful. Although several NPRS systems have been successfully modified to produce novel products, these modifications have all resulted in significant decreases in product synthesis. This is perhaps due to the highly dynamic nature of NPRS enzymes, and to the effect that the relatively unstudied module-linker sequences have on action. Another problem is that, in addition to the A domain, the less well characterised C domain may also influence domain selectivity.[27]
3.8 - NRP/PK conclusions
Even though Polyketides and Non-ribosomal peptides are structurally unrelated, their biosynthetic pathways are highly comparable, and as such they are often discussed in tandem. Both of these natural products are highly medically useful, and are therefore very well studied and understood. Both classes of synthetase produce products highly diverse in size and structure, and yet there is a high degree of homology between the enzymes themselves. These synthesis pathways have been finely tuned through multiple rounds of evolution to produce a vast array of products from a limited monomer subset. Differences in domain composition have allowed for the capability of both classes of synthetase to produce a massive range of products. It would be almost impossible for humans to match the vast array of complex and diverse scaffolds that microbes are able to produce through different combinations of the PKS/NPRS domains and modules. It is equally difficult for synthetic laboratory drug production methods to match the high degree of product specificity that natural microbial synthetases display; especially with regard to the orientation of stereogenic centres which are highly regulated by certain module domains. However, it is possible that as our understanding of these biosynthetic pathways increase we may be able to harness these pathways to tailor existing drugs or even to produce novel scaffolds.
4 - THE WAY AHEAD: DISCUSSIONS AND CONCLUSIONS
The demand for novel antibiotics is clear and urgent; to combat the increasing number of multi-drug resistant bacteria it is imperative that more research be undertaken to discover/develop new drugs. Although this is undeniable, there is some contention as to whether this research should focus on the discovery of natural products, or on synthetic chemistry. In this section we will discuss the advantages, disadvantages and future of both.
4.1 - Natural products
As has been shown throughout this essay, microbes are capable of producing a vast array of antimicrobial natural products from a relatively limited simple subset of monomers with an impressive degree of accuracy and innovation. Throughout the last century, this diverse array of chemical compounds has provided the basis for most clinically available antibiotics. The complexity and diversity of these natural scaffolds is such that it would be nearly impossible for combinatorial chemistry libraries to match the scope of compounds produced by microbes.
However, despite the obvious impact of naturally sourced antibiotics, in recent decades, discovery of novel natural antibiotics has slowed. It is certainly not the case that all natural antibiotics have been discovered; indeed, it is estimated that only 1-3% of all antibiotics produced by the Streptomycetes (a genus in the order Actinomycetes, the most prolific producers of antibacterials) have so far been identified.[29] However, in order to increase the likelihood of discovery several courses of action must be taken. Namely increasing the scope of species under investigation, developing new culturing techniques, and developing new assays.
Study of new microbes obtained from original sources has already yielded significant results. The examination of microbes from the Sea of Japan led to the discovery of the polyketide antibiotic Abyssomycin (produced by the actinomycete Verrucosispora maris), which inhibits the bacterial folate-synthesis pathway. Analysis of bacteria of forest soil in Italy has also uncovered two new genera of Actinomycetes, whose genomes indicate a great potential for production of both polyketides and non-ribosomal peptides.[29]
The development of new culturing techniques will also provide a productive way ahead; rRNA studies indicate that only 1% of all bacterial species are culturable using standard culturing methods, broadening this spectrum would certainly increase the chances of discovery.[29]An excellent example of the importance of this is the discovery of the new antibiotic teixobactin, whose productive bacteria was first cultured using the novel iChip device, designed to culture soil bacteria. This device isolates single bacteria into individual wells, which can then be placed back into the sample soil, allowing the trapped microbes to grow and replicate in their natural environment. The growth recovery of microbes cultured using iChip is approximately 50% (compared to the 1% culturable using normal petri dish methods).[14]
4.2 - Alternatives to natural products
The impact that natural products have had on the pharmaceutical industry is clear; approximately 60% of drugs in use today were derived from natural sources. Even though natural products have provided the basis for many drugs, it is common practice for these natural organic compounds to be chemically altered to improve features such as solubility, stability and efficacy prior to industrial production.[1] The penicillin class of antibiotics are an excellent example of this; since the discovery of the original Penicillin G, this molecule has been modified to produce multiple variants with increased efficacy, the ability to evade resistance mechanisms, and an increased target spectrum.
While the derivatives of natural products had dominated the pharmaceutical industry for much of the Twentieth Century, from the 1990’s, a greater focus has been placed on combinatorial chemistry and high throughput screening (HTS). HTS relies on the use of automated robotic platforms to assay the effect of members of compound libraries synthesised using solid phase methods, with the goal of identifying potential drugs. Although it should be noted that natural products are tested for drug activity using the same screening methods, the introduction of HTS caused a shift towards the study of synthetic chemical libraries and caused many pharmaceutical companies to downsize natural product research.[2] Some believe that the decrease in discovery of novel drugs in recent decades can be, in part, attributed to this switch.[1]
Despite the belief that combinatorial chemistry would be a major source for novel drugs and compounds, only one New Chemical Entity produced in this manner (the kinase inhibitor Sorafenib) was accepted for use by the FDA. And despite this focus on human-designed over natural products, still many of the drugs in use are modifications of naturally produced scaffolds. (A notable exception to this trend is the quinolone class of antibiotics, which were first synthesised 40 years ago, and are still used today.[2])The primary disadvantages of combinatorial chemical libraries is their relative simplicity compared with natural scaffolds, and the fact that optimisation of synthetic molecules requires far more effort than modifying natural products which have been optimised by evolutionary pressure.
5 - SUMMARY
The biosynthetic pathways that have developed in microorganisms over the millions of years of directed evolution have been fine tuned to produce a vast array of complex and diverse bioactive compounds from a simple subset of ubiquitous monomers. These complex synthesis enzymes can not only control the order and structure of the polymers produced from these monomers but are also capable of mediating small chemical changes which attenuate the bioactivity of the compound. Due to the modular nature of these enzymes, the range of possible products which can be biosynthesised is almost infinite in their size and diversity. It is currently not possible for this level of complexity and diversity to be matched by synthetic chemistry.
Rather than continuing to focus on HTS of combinatorial chemistry libraries, a shift of research focus onto the discovery of novel scaffolds from new sources, and the harnessing of these biosynthetic pathways may provide a more productive future for medicine discovery and production. The recent discovery of Teixobactin using a novel culturing technique may provide a model for the way ahead.
REFERENCES
[1] Ji, H.-F., Li, X.-J., & Zhang, H.-Y. (2009). Natural products and drug discovery. Can thousands of years of ancient medical knowledge lead us to new and powerful drug combinations in the fight against cancer and dementia? EMBO Reports, 10(3), 194–200. http://doi.org/10.1038/embor.2009.12
[2] Dias, D. A., Urban, S., & Roessner, U. (2012). A Historical Overview of Natural Products in Drug Discovery. Metabolites, 2(2), 303–336. http://doi.org/10.3390/metabo2020303
[3] Hanson, J. R. (2003). Natural products: the secondary metabolites. Cambridge: Royal Society of Chemistry.
[4] Clardy, J., Fischbach, M. A., Walsh, C. T. (2006). New antibiotics from bacterial natural products Nature Biotechnology 24, 1541–1550 (2006) doi:10.1038/nbt1266
[5] Basmadjian, C., Zhao, Q., Bentouhami, E., Djehal, A., Nebigil, C. G., Johnson, R. A., … Désaubry, L. G. (2014). Cancer wars: natural products strike back. Frontiers in Chemistry, 2, 20. http://doi.org/10.3389/fchem.2014.00020
[6] Aminov, R. I. (2010). A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Frontiers in Microbiology, 1, 134. http://doi.org/10.3389/fmicb.2010.00134
[7] Diminic, J., Starcevic, A., Lisfi, M. et al. (2014) Evolutionary concepts in natural products discovery: what actinomycetes have taught us J Ind Microbiol Biotechnol 41: 211. https://doi.org/10.1007/s10295-013-1337-8
[8] Ventola, C. L. (2015). The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharmacy and Therapeutics, 40(4), 277–283.
[9] Holland, A., & Kinnear, S. (2013). Interpreting the Possible Ecological Role(s) of Cyanotoxins: Compounds for Competitive Advantage and/or Physiological Aide? . Marine Drugs, 11(7), 2239–2258. http://doi.org/10.3390/md11072239
[10] Encyclopedia of Microbiology (Third Edition) (3rd ed., pp. 318-332). (2009).
[11] Gomes, E. S., Schuch, V., & de Macedo Lemos, E. G. (2013). Biotechnology of polyketides: New breath of life for the novel antibiotic genetic pathways discovery through metagenomics. Brazilian Journal of Microbiology, 44(4), 1007–1034.
[12] Chopra, I., & Roberts, M. (2001). Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiology and Molecular Biology Reviews, 65(2), 232–260. http://doi.org/10.1128/MMBR.65.2.232-260.2001
[13] National Center for Biotechnology Information. PubChem Compound Database; CID=12560, https://pubchem.ncbi.nlm.nih.gov/compound/12560 (accessed Jan. 29, 2018).
[14] Ling, L. L. et al (2015). A new antibiotic kills pathogens without detectable resistance. Nature, 517(7535), 455-459. doi:10.1038/nature14098
[15] Jin, K. et al (2016). Total synthesis of teixobactin. Nature Communications, 7, 12394. doi:10.1038/ncomms12394
[16] Breinbauer R., Mentel M. (2010) Combinatorial Chemistry and the Synthesis of Compound Libraries. In: Roque A. (eds) Ligand-Macromolecular Interactions in Drug Discovery. Methods in Molecular Biology (Methods and Protocols), vol 572. Humana Press, Totowa, NJ
[17] Khosla, C., Herschlag, D., Cane, D. E., & Walsh, C. T. (2014). Assembly Line Polyketide Synthases: Mechanistic Insights and Unsolved Problems. Biochemistry, 53(18), 2875–2883. http://doi.org/10.1021/bi500290t
[18] Wu, J., Zhang, Q., Deng, W., Qian, J., Zhang, S., & Liu, W. (2011). Toward Improvement of Erythromycin A Production in an Industrial Saccharopolyspora erythraea Strain via Facilitation of Genetic Manipulation with an Artificial attBSite for Specific Recombination. Applied and Environmental Microbiology, 77(21), 7508–7516. http://doi.org/10.1128/AEM.06034-11
[19] Strieker, M., Tanović, A., & Marahiel, M. A. (2010). Nonribosomal peptide synthetases: structures and dynamics. Current Opinion in Structural Biology, 20(2), 234-240. doi:10.1016/j.sbi.2010.01.009
[20] Challis, G. L., & Naismith, J. H. (2004). Structural aspects of non-ribosomal peptide biosynthesis. Current Opinion in Structural Biology, 14(6), 748–756. http://doi.org/10.1016/j.sbi.2004.10.005
[21] Walsh, C. T. (2016). Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Natural Product Reports, 33(2), 127-135. doi:10.1039/c5np00035a
[22] Shaligram, N. S., & Singhal, R. S. (2010). Surfactin – A Review on Biosynthesis, Fermentation, Purification and Applications. Food Technol. Biotechnol., 48(2), 119-134.
[23] Bruner, S., Weber, T., Kohli, R., Schwarzer, D., Marahiel, M., Walsh, C., & Stubbs, M. (2002). Structural Basis for the Cyclization of the Lipopeptide Antibiotic Surfactin by the Thioesterase Domain SrfTE. doi:10.2210/pdb1jmk/pdb
[24] Du, L., Sánchez, C., & Shen, B. (2001). Hybrid Peptide–Polyketide Natural Products: Biosynthesis and Prospects toward Engineering Novel Molecules. Metabolic Engineering,3(1), 78-95. doi:10.1006/mben.2000.0171
[25] Letzel, A., Pidot, S. J., & Hertweck, C. (2013). A genomic approach to the cryptic secondary metabolome of the anaerobic world. Nat. Prod. Rep., 30(3), 392-428. doi:10.1039/c2np20103h
[26] Zhang, J., Yan, Y., An, J., Huang, S., Wang, X., & Xiang, W. (2015). Designed biosynthesis of 25-methyl and 25-ethyl ivermectin with enhanced insecticidal activity by domain swap of avermectin polyketide synthase. Microbial Cell Factories, 14(1). doi:10.1186/s12934-015-0337-y
[27] Bozhüyük, K. A., Fleischhacker, F., Linck, A., Wesche, F., Tietze, A., Niesert, C., & Bode, H. B. (2017). De novo design and engineering of non-ribosomal peptide synthetases. Nature Chemistry. doi:10.1038/nchem.2890
[28] Walsh, C. T., & Wencewicz, T. A. (2013). Prospects for new antibiotics: a molecule-centered perspective. The Journal of Antibiotics, 67(1), 7-22. doi:10.1038/ja.2013.49
[29] Bayly, C., & Yadav, V. (2017). Towards Precision Engineering of Canonical Polyketide Synthase Domains: Recent Advances and Future Prospects. Molecules, 22(2), 235. doi:10.3390/molecules22020235
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biology"
Biology is the scientific study of the natural processes of living organisms or life in all its forms. including origin, growth, reproduction, structure, and behaviour and encompasses numerous fields such as botany, zoology, mycology, and microbiology.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: