Structural Insights for Drugs Developed for Phospholipase D Enzymes
Info: 8531 words (34 pages) Dissertation
Published: 17th Feb 2022
Tagged: Biomedical Science
ABSTRACT
In recent years human phospholipase D enzymes (PLD1 and PLD2 isozymes) have emerged as drug targets for various diseases such as cardiovascular disease, cancer, infectious diseases and neurodegenerative conditions such as Alzheimer’s and Parkinson’s disease. The interest in PLD as a drug target is due to the fact that PLD enzymes belong to a superfamily of phospholipases that are essential to intracellular and extracellular signaling. Many bioactive lipid signaling molecules are generated by these enzymes including phosphatidic and lysophosphatidic acid, arachidonic acid, and diacylglycerol (DAG). More specifically PLD’s are part of one pathway that generates phosphatidic acid which is a precursor to many lipids in the intracellular de novo pathway. The lipids produced from PA regulate many cellular events considered hallmarks of pathogenesis in cells; including proliferation, migration, invasion, angiogenesis, and vesicle transport. Hence, human PLD is a valid target for a variety of drug therapies.
An enormous amount of cellular–based work has been done in the last decade to illustrate the essential role of PLD in cancer growth and development, and more recently neurodegenerative diseases and infectious diseases. A survey of structure-based drug design studies for PLD enzymes is discussed, in addition, structural investigation utilizing the available three dimensional coordinates of PLD and recent potent PLD isozyme specific inhibitors via in silico docking and comparison with enzyme-based assays may provide valuable insights into the mode of binding by drugs designed to inhibit PLDs.
Keywords: Binding mode, allosteric, docking studies, IC50, and drug discovery.
Phospholipase D (PLD) enzymes have been defined as a class of phosphodiesterases that hydrolyze phosphatidylcholine and other amine-containing glycerophospholipids to generate phosphatidic acid (PtdOH) and a free head group [1-8]. The human phospholipase enzymes hPLD1 and hPLD2 have been extensively studied as “classical” PLD’s in that they cleave phosphatidylcholine to generate free choline and PtdOH (Figure 1A) and have the conserved sequence of amino acids (H-X-K-X4-D-X6-G-G/S) known as the HKD motif in their active sites. The reaction mechanism for PLD’s of this type is very similar. However, there also exists some PLD family members that lack the HKD motif that generate PtdOH [6, 9].
In addition, many PLD enzymes also catalyze a transphosphatidylation reaction in which short chain primary alcohols compete with water as a nucleophile to generate products known as phosphatidyl alcohols [1}, a reaction which has the effect of contributing to a decrease of intracellular pools of PtdOH in the presence of primary alcohols. Although PLD has traditionally been thought of as the source of PtdOH a potent signaling lipid that that effects membrane structure, PtdOH is also central to the synthesis of other bioactive compounds. As shown in Figure 1B, there are several biosynthetic pathways which generate PtdOH [1, 10]. A vast amount of literature has been written on both the bioactive properties of PtdOH in terms of cell signaling and vesicle transport and PLD protein-protein interactions for neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease[10], cellular survival and proliferation (cancer) involving PLD modulation/influence on many signaling pathways [1-8, 10-11].
The development and diversity of PLD-specific inhibitors
In response to the many physiological studies that have been done linking PLD to pathological conditions, PLD specific inhibitors have been developed over the last several years. The main targets for drug development have been hPLD1 and hPLD2 isozymes, the endocannabinoid regulator N-acyl phosphatidylethanolamine-specific phospholipase D (NAPE-PLD) and bacterial PldA [1, 12-16]. Target validation of small molecule inhibitors of PLD began with phosphate mimics such as tungstate and vanadate then evolved onward to natural products such as SCH420789 and calphostin C [1] . However, due to lack of specificity these molecules did not develop into viable inhibitors of PLD [1]. Finally in 2007, a breakthrough occurred that catalyzed current research into PLD inhibitors with the initial discovery that halopemide , an antipsychotic agent used in humans also inhibits hPLD2 at IC50 of 200 nM [1, 17]. From the original halopemide molecule 14 analogues were developed one of which is now known as FIPI (5-fluoro-2-indoyly des-chlorohalopemide (Figure 2A).
Although originally reported for hPLD2, halopemide and FIPI and analogues are actually dual hPLD1 and hPLD2 inhibitors. The comparative cellular and biochemical IC50’s of halopemide and FIPI are shown in Table 1 [1]. Many biochemical and cellular-based studies were done on these molecules for target validation some studies which were valuable as highly selective PLD1 and PLD2 inhibitors were developed. [18- 26]. However halopemide and FIPI compounds have > 30 off target activities (reactive at many biogenic amine receptors) as reported by Brown, Thomas and Lindsley: “expected for atypical antipsychotics that have G-protein coupled receptors (GPCR)-privileged structures as a core motif” [1]. Clearly a new strategy of small molecule optimization needed to be employed with improved ancillary pharmacology and drug metabolism and pharmacokinetic characteristics (DMPK–referring to absorption, distribution, metabolism and excretion of a drug) [1,13].
By combining the methods of diversity-oriented synthesis with iterative parallel synthesis, and“magic methyl” effect, a second generation inhibitor known as VU0359595 with only 7 off-target activities was generated based on halopemide and FIPI [ Figure 2B}. Interestingly, this inhibitor VU0359595 exhibited selectivity towards PLD1 with an IC50 of 3.7 nM compared with PLD2 IC50 of 6,400 nM with much improved ancillary pharmacology and DMPK profile [ 1, 13, 21]. The search for a potent PLD2 inhibitor did not yield results within the piperidine benzimidazolone core analogue. However, an N-phenyl triazaspirone congener was discovered to show a preference for PLD2 with an IC50 of 110 nM versus PLD1 IC50 of 1,000 nM [1, 22].
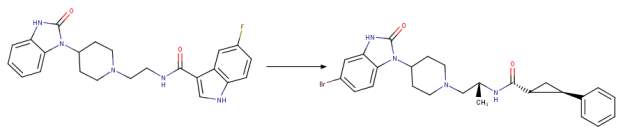
After a rigorous lead optimization project focusing on the N-phenyl triazaspirone congener, it was discovered that addition of a fluorine atom in the 3rd position of the N-phenyl moiety produced much improved PLD2 selectivity VU0364739 with PLD2 IC50 of 20 nM. It is fascinating to note that on the VU0354739 compound that just the addition of a fluorine atom could drop thePLD2 IC50 fivefold and induce a 75 fold difference in potency when compared with IC50.for PLD1 of 1,500 nM [22] Interestingly, a potent dual inhibitor for PLD1 and PLD2 was prepared by addition of an (S)-methyl group into the N-phenyl triazaspirone congener, that showed a dramatic drop in IC50 for PLD1 [Figure 2C]. The IC50 for PLD1 dropped from 1000, nM to 25 nM and PLD2 specificity remained reasonable with an IC50.of 140 nM. It seems that the small structural change of adding the (S)-methyl group acted like a “molecular switch” previously observed in allosteric GPCR ligands [1, 21-24].
The third generation of phospholipase D2 selective and dual phospholipase D1-phospholipase D2 inhibitors within the triazaspirone series were most likely prepared to further the potency of the dual inhibitors and decrease the off-target activities and further improve the DMPK profile. This work led to the synthesis of the compound ML298 which showed an IC50 of 355 nM for PLD2 and ~ 20 M for PLD1. The incorporation of the (S)-methyl group that was used in the piperidine benzimidazolone series and the addition of a bromine (instead of 2 fluorine atoms) resulted in compound ML299 (Figure 2D). ML299 exhibited enhanced PLD1 inhibition 250 fold with IC50 of 6 nM and PLD2 IC50 of 320 nM. Interestingly ML298 was restricted peripherally but ML299 could penetrate the central nervous system [1, 24].
Next a variety of non-N-aromatic moieties (not bound to nitrogen) were evaluated in order to prepare a PLD2 inhibitor as potent as ML298 but also CNS penetrant [24]. ML395 (a pyridylmethyl congener) was synthesized. ML395 exhibited PLD1 IC50 > 30,000 nM and PLD2 IC50 380 nM. In addition to this profound selectivity for PLD2, ML395 exhibited an excellent DMPK profile and high CNS penetration (important for inhibiting PLD in neurodegenerative diseases). This was the first PLD inhibitor with no off-target activities reported [25]. A whole series of isozyme-selective potent PLD inhibitors have been developed. Since the halopemide-derived and novel triazaspirone-based series of PLD inhibitors were specific for mammalian PLDs as far as they were tested, a new campaign began for the development of a universal PLD inhibitor.
More specifically, PLD inhibitors developed did not inhibit Pseudomonas aeruginosa PldA and the endocannabinoid regulator NAPE-PLD [1, 15]. Selective oestrogen receptor modulators (SERMS) are compounds used in the treatment of oestrogen-receptor-positive breast cancer that have been reported to inhibit mammalian PLD enzymes in the low micromolar range. [1, 27-29]. Examples of such inhibtors are: Raloxifene, Tamoxifen, 4-hydroxytamoxifen and Desketoraloxifene.
Desketoraloxifene is the most potent PLD1, PLD2 and PldA inhibitor and became the focus to develop the first universal PLD inhibitor with analogues designed to abolish SERM activity. [1, 15]. The first universal PLD inhibitor developed was devoid of the 6-OH moiety necessary for oestrogen receptor binding and anti-proliferation action. The compound contains a N,N-dimethylamino moiety which is known to reduce SERM activity (Figure 3) [1, 30]. The PLD universal inhibitor exhibited an IC50’s of 4.7 M for PLD1, 7.1 M for PLD2, 0.5 M PldA and 67 M for NAPE PLD [15]. Further studies are in progress to develop more potent analogues for this first universal PLD inhibitor [1].
A survey of plausible binding modes for PLD inhibitors in the active site and allosteric sites
Since the PLD isozymes were so selective and contained “molecular switches”, researchers began to investigate the mechanism by which these compounds inhibit PLDs [1]. Interestingly PLD inhibitors have similarity to AKT piperdine benzimidazolone inhibitors of AKT [1] . Inhibitors of AKT have two binding modes: catalytic domain binding and pleckstrin homology (PH) domain binding [31]. It is well established that hPLD1 and hPLD2 share: 1) 2 HxKxxxxD (HKD) motifs for catalysis, 2) a phox (PX) consensus sequence, a pleckstrin homology (PH) domain and PIP2 binding site [10,32 ]. Investigations have been done with enzyme kinetics [13, 21-26 ], and followed up with backscattering interferometry technique to determine if binding modes of PLD inhibitors are allosteric [31, 33].
Early studies with halopemide and FIPI assays using a PLD1.311 truncated construct (deletion to part of the pleckstrin homology domain) did not show large reduction of PLD activity [13] (but did show significant decrease with VU0395595 and VU0364739) [1]. This implies two possible binding modes for PLD inhibitors and was confirmed using backscattering interferometery technique [34] that there were 2 sites of hPLD1 protein for VU0359595: one with high affinity and one with low affinity [35]. There is also strong biochemical evidence for two modes of binging catalytic and allosteric binding using FIPI in both the catalytic site and the allosteric phosphoinositide-binding pocket. FIPI interacted with S757 of PLD2 within the PIP2 pocket, as well as other PLD isozymes [1, 31,36]. The PLD2 specific inhibitor VU0364739 interacted with two different PLD2 sites in the HKD (S757 or S648) and an allosteric site R210 or R212 which is the phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) binding domain.
In addition, F244-L245-L246 of PLD2 forms a hydrophobic pocket in the PH domain which is also found in AKT at the allosteric binding site for similar inhibitors [31, 36]. Taken together, a three dimensional crystal structure of hPLD1 and PLD2 isozymes in complex with inhibitors described herein is paramount to confirming these very meticulous and convincing biochemical binding and kinetic studies.
In the absence of the three dimensional structures of hPLD1 and hPLD2, the coordinates of the Streptomyces species pmf phospholipase D (PLDpmf) was used to investigate plausible binding models of human PLD inhibitors using in silico docking experiments. The PLDpmf species has high sequence homology with hPLD1 as previously described [37]. However, before docking studies were done amino acid sequence analysis was done comparing hPLD1 and hPLD2 to PLDpmf . Utilizing alignment tool (lalign) local regions of the amino acid sequences of hPLD1 and hPLD2 align with PLDpmf in the region of the HKD repeats and PX and PH domains [38].
More specifically regions of hPLD1 and PLDpmf aligned with 28-44% identity and 55-77% similarity. The alignment scores are lower for PLDpmf and hPLD2 at 23-28% identity and 52-60% similarity. Utilizing SMART sequence analysis tools (simple modular architecture research tool) details of the amino acid range of the well conserved domains was determined. More specifically, The PX (phox) domain found in proteins that are involved in vesicle trafficking and cell signaling, in hPLD1 is between amino acids 81-212 and the PH domain (where allosteric binding occurs of selective PLD inhibitors) is between amino acids 219-328 [39, 40]. Interestingly S167 from PLDpmf aligns with S757 of hPLD2, mentioned above in the allosteric studies of binding modes for PLD inhibitors and several of the hPLD1 and hPLD2 inhibitors successfully docked in that region. Taken together careful inspection of docking results and sequence alignments “matches” may be found using the PLDpmf 3D coordinates as a guide to predict plausible binding modes for the hPLD1 and hPLD2 inhibitors.
After comparing several docking programs results with a quick shape descriptor algorithm [41], and the actual IC50’s with many of the known hPLD1 and hPLD2 inhibitors, Dock6.8 [42] was chosen as the docking program for this study because the binding constants reported in the docking results were within ~30% error of the actual IC50’s reported in the literature. As shown in Figure 4A, PDB code 1V0Y [43] FIPI is docked in the active site of the
PLDpmf structure made up of amino acids: K450, N465, Y467, W168, L90, N187, L172, H448, and H170. Interestingly H756 of hPLD2 aligns with H170 as do the other histidines and lysines in the active site. As shown in the surface rendering of 1V0Y active site with docked FIPI in Figure 4B, there is a hydrophobic pocket directly “above” W168 which seems to stabilize the docked FIPI inhibitor. Amino acids from this hydrophobic pocket such as W237 are highly conserved and several PLD inhibitors docked into this hydrophobic pocket blocking access to the active site but not directly engaging active site residues in binding.
From docking observations, there is no obvious hydrogen bonding network for the fluorine atom in the FIPI inhibitor. The calculated binding constant for docking FIPI in the active site of 1V0Y was 0.0125 mM or 12.5 nM. Although “blind” docking experiments were attempted and FIPI did “fit” into a plausible binding pocket in PLDpmf adjacent to the active site, the bonding constant was 550 nM and so those results are not considered plausible based on the data of the actual IC50’s. Interestingly in Figure 5, when the short chain PtdOH product co-crystallized with the PLDpmf is left in the active site of 1V09, the ML299 inhibitor (Figure 2D) binds to a plausible “allosteric site” below the active site. Interestingly, S167 which aligns with PLD2 S757 implicated in biochemical allosteric binding studies of PLD2 inhibitors [1] is in close proximity to the bromine atom on this compound (Figure 5).
The three dimensional structure of the human NAPE-PLD has been solved and was also utilized in these docking studies to observe the PLD universal inhibitor binding in either the active site or a plausible binding pocket out of the active site in allosteric binding mode. Using PDB coordinates 4QN9 [44], the universal PLD inhibitor (Figure 3) was docked in the active site of 4QN9 as shown in Figure 6A. The binding constant for this docking observation is 105 M (the actual IC50 is 67 M). The docking observed suggested that T258 hydroxyl group was in close enough proximity to the inhibitor to hydrogen bond to the nitrogen on the inhibitor.
Y188 and R257 are in close enough proximity to predict hydrogen bonding to the inhibitor. Once again the majority of the predicted interactions are hydrophobic. Within the NAPE-PLD active site, the docked universal inhibitor appears stabilized mainly by hydrophobic interactions with W254, Y188, W218, and W254. As shown in Figure 6B, the NAPE-PLD docked inhibitor is observed to be in a plausible position to bind to a hydrophobic pocket adjacent to the active site, which may in effect be blocking access to the active site for the natural substrate of NAPE-PLD.
PLD and infectious diseases: Docking of PLDpmf and human AKT
Recent research has implicated PLD in infectious diseases. More specifically the role of hPLD2 in host cell infection with influenza virus may be to provide PtdOH for recruitment of signaling molecule AKT to the cellular membrane where AKT would phosphorylate downstream effector Beclin 1 at S295 promoting autophagy [1]. Interestingly in this illustration of a recent publication [1] it was shown that hPLD2 and AKT are engaging in a specific, direct protein-protein interaction [1,45] Therefore it is not just the curvature of membranes by PtdOH to promote AKT binding but a specific interaction of the two proteins.
To investigate this idea further, the PDB coordinates for AKT 5KCV [46] were downloaded and docked with PLDpmf 1V0Y in a protein-protein docking program called ZDOCK [47]. As shown in Figure 7, the PLDpmf (magenta) forms a convincing heterodimer with human AKT structure (blue). The ligands were omitted during the docking and then put in as a reference for visualization. The short chain PtdOH is shown in the PLDpmf active site and the allosteric inhibitor RQ092 (3-(3-(4-(1-aminocyclobutyl)phenyl)5-phenyl-3H-imidazo[4,5-6]pyridine-2-4)pyridine-2-amine) is shown in AKT structure [46]. The interface between the two docked structures has convincing amino acid interactions.
More specifically, from the docking results, the “top” of the figure at the interface PLDpmf S135 interacted with ART K154, PLDpmf K136 was with hydrogen bonding distance of ARK T435 and R436. A hydrophobic pocket interaction is observed between PLDpmf I133 and I233 and AKT L155 and F438. Approaching the active site of PLDpmf S235 interacts with Q428 on AKT. There is a helix of AKT with hydrophobic residues that align well with the crystallographic PtdOH product—F236/237, L239 and V244 on AKT are well positioned to interact with PtdOH. A125 on loop near PLDpmf active site is nearby. Towards the “bottom” of the interface PLDpmf S291 and AKT Q352 are observed within hydrogen bonding distance and PLDpmf R382 and AKT N351 are within hydrogen bonding range. Although more sequence analysis needs to be done to “match” these PLDpmf residues with hPLD1 and hPLD2, since as mentioned above PLDpmf has local regions of reasonably high identity and homology with hPLD1 and hPLD2, the docking results are very intriguing.
In conclusion, the work herein highlights and summarizes some of the most promising PLD inhibitors recently developed and characterized through some elegant synthesis regimes and biochemical studies. In addition, docking studies were done to gain insight as to the binding modes of these molecules in three dimensional structures available from the protein data bank Rutgers Consortium of Structural Biology (RCSB). However as any self-respecting/aspiring crystallographer knows from a structural point of view, unless there is a co-crystallized ligand or small molecule with reasonable B factors docked or built into a difference density with sigma cutoff of 2.0 or higher, one cannot be certain of the actual positioning of the molecules being analyzed.
Hopefully, at the time of the writing of this work herein some energetic group of researchers is well along the way to solving the three dimensional structures of both hPLD1 and hPLD2, perhaps soaked or co-crystallized with some of the potent inhibitors described herein, to be deposited into the PDB for all to see and use for analysis and confirmation of kinetic and biochemical binding studies.
References
1. Brown, A.H., Thomas, P.G., Lindsley, C.W., Targeting phospholipase D in cancer, infection, and neurodegenerative disorders. Nature Reviews Drug Discovery, 16, 351-367 (2017).
2. Ponting, C. P. & Kerr, I. D. A novel family of phospholipase D homologues that includes phospholipid synthases and putative endonucleases identification of duplicated repeats and potential active site residues. Protein Sci. 5, 914–922 (1996).
3. Sato, T., Hongu, T., Sakamoto, M., Funakoshi, Y. & Kanaho, Y. Molecular mechanism of N-formyl-methionyl-leucyl-phenylalanine-induced superoxide generation and degranulation in mouse neutrophils:phospholipase D is dispensable. Mol. Cell. Biol. 33, 136–145 (2013).
4. Bruntz, R. C., Lindsley, C. W. & Brown, H. A.Phospholipase D and phosphatidic acid signaling pathways as targets for therapeutics: roles in cancer. Pharmacol. Rev. 66, 1033–1079 (2014).
5. Peng, X. & Frohman, M. A. Mammalian phospholipase D physiological and pathological roles. Acta. Physiol. (Oxf). 204, 219–226 (2012).
6. Selvy, P. E., Lavieri, R. L., Lindsley, C. W. & Brown, H. A.Phospholipase D: enzymology, signaling, and chemical modulation. Chem. Rev. 111, 6064–6119 (2011).
7. Oude Weernink, P. A., Han, L., Jakobs, K. H. & Schmidt, M. Dynamic phospholipid signaling by G protein-coupled receptors. Biochim. Biophys. Acta. 1768, 880–900 (2007).
8. DeYonker, N. J. & Webster, C. E. Phosphoryl transfers of the phospholipase D superfamily: a quantum mechanical theoretical study. J. Am. Chem. Soc. 135, 13764-13774 (2013).
9. Yang, H., Roberts, MF. Phosphohydrolase and transphosphatidylation reactions of two Streptomyces phospholipase D enzymes: covalent versus noncovalent catalysis. Protein Sci. 12, 2087-2098.
10. Oliveira, T.G., Paolo, G.D., Phospholipase D in brain function and Alzheimer’s disease. Biochim. Biophys. Acta., 1801,799-805 (2010).
11. Chang, S, Torok, B, Stieglitz, K.A. The role of phospholipase D enzyme(s) in modulating cell signaling: Implications for Cancer Drug Development. CBC, 10(2),124-130 (2014).
12. Kulkarni A, Quang P, Curry V, Keyes R, Zhou W, Cho H, Baffoe J, Török B, Stieglitz K. 1,3-disubstituted-4-aminopyrazolo [3, 4-d] pyrimidines, a new class of potent inhibitors for phospholipase D. Chem Biol Drug Des. 84, 270-281.
13. Scott, S. A. et al. Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat. Chem. Biol. 5, 108–117 (2009).
14. Scott, S. A., Mathews, T. P., Ivanova, P. T., Lindsley, C. W. & Brown, H. A. Chemical modulation of glycerolipid signaling and metabolic pathways. Biochim. Biophys. Acta. 1841, 1060–1084 (2014).
15. Scott, S. A. et al. Discovery of desketoraloxifeneanalogs as inhibitors of mammalian, Pseudomonas aeruginosa, and NAPE phospholipase D enzymes. ACS Chem. Biol. 10, 421–432 (2015).
16. Egertová, M., Simon, G. M., Cravatt, B. F. & Elphick, M. R. Localization of N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) expression in mouse brain: a new perspective on N-acylethanolamines as neural signaling molecules. J. Comp. Neurol. 506, 604–615 (2008).
17. Monovich, L. et al, Optimization of halopemide for phospholipase D2 inhibtion. Bioorg. Med. Chem. Lett. 17, 2310-2311 (2007).
18 Loonen, A. J., Soe-Agnie, C. J. & Soudijn, W. Effects of halopemide on GABA receptor binding, uptake and release. Brain Res. 210, 485–492 (1981).
19 Loonen, A. J. & Soudijn, W. Halopemide, a new psychotropic agent. Cerebral distribution and receptor interactions. Pharm. Weekbl. Sci. 7, 1–9 (1985).
20 Su, W. et al. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol. 75, 437–446 (2009).
21. Lewis, J. A. et al. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I:impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg. Med. Chem. Lett. 19, 1916–1920 (2009).
22. Lavieri, R. et al. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part II:identification of the 1,3,8-triazaspiro[4,5]decan-4-one privileged structure that engenders PLD2 selectivity. Bioorg. Med. Chem. Lett. 19, 2240–2243 (2009).
23. Lavieri, R. R. et al. Design, synthesis and biological evaluation of halogenated N-(2-(4-oxo-1-phenyl-1,3,8-triazasprio[4.5]decan-8-yl)ethyl)benzamides: discovery of an isoform-selective small molecule phospholipase D2 (PLD2) inhibitor. J. Med. Chem. 53, 6706–6719 (2010).
24. O’Reilly, M. C. et al. Development of dual PLD1/2 and PLD2 selective inhibitors from a common 1,3,8-triazaspiro[4.5]decane core: discovery of ML298 and ML299 that decrease invasive migration in U87-MG glioblastoma cells. J. Med. Chem. 56, 2695–2699 (2013).
25. O’Reilly, M. C. et al. Discovery of a highly selective PLD2 inhibitor, VU0468809 (ML395): a new probe with improved physiochemical properties and broad spectrum antiviral activity against influenza strains. ChemMedChem. 9, 2633–2637 (2014).
26 O’Reilly, M. C., Scott, S. A., Brown, H. A. & Lindsley, C. W. Further evaluation of novel structural modifications to scaffolds that engender PLD isoform selective inhibition. Bioorg. Med. Chem. Lett. 24, 5553–5557 (2014).
27. Cummings, S. R. et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 281, 2189–2197 (1999).
28. Plowman, P. N. Tamoxifen as adjuvant therapy in breast cancer. Current status. Drugs 46, 819–833 (1993).
29. Eisen, S. F. & Brown, H. A. Selective estrogen receptor modulators differentially regulate phospholipase D catalytic activity in ER-negative breast cancer cells. Mol. Pharmacol. 62, 911–920 (2002).
30. Grese, T. A. et al. Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc. Natl Acad. Sci. USA 94, 14105–14110 (1997).
31. LindseyC.W., et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 15, 761-764 (2005).
32. Oliviera and:G.M. Jenkins, M.A. Frohman, Phospholipase D: a lipid centric review, Cell. Mol.Life Sci. 62 (2005) 2305–2316.].
33. DeFeo-Jones, D. et al. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol. Cancer Ther. 4, 271-279 (2005).
34. Bornhop, D.J., Baksh, M, Finn, M.G. & Kussrow, A. Backscaterring interferometric analysis of membrane materials, US Patent 20130040306 (2013).
35. Lavieri, R.R. Synthesis, Development and Biochemical Characterization of Small Molecule, Isoform-Selective Phospholipase D Inhibitors and Photoreactive Probes. PhD. Thesis, Vanderbilt Univ. (2014).
36. Ganesan, R., Mahankali, M., Alter, G.& Gomez-Cambronero, J. Two sites of action for PLD2 inhibitors: the enzyme catalytic center and an allosteric phosphoinositide binding pocket. Biochim. Biophys. Acta. 1851, 261-272 (2015).
37. Leiros, I., Secundo, F, Zambonelli, C., Servi, S. Hough, E. The first crystal structure of a phospholipase D. Structure, 8, 655-667. (2001).
38. http://embnet.vital-it.ch/software/LALIGN_form.html (last accessed 6/3/17)
39. Schultz, J., Milpetz, F., Bork, P., Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 95, 5857-5864. (1998).
40. Letunic, I, Doerks, T, Bork,P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res 43, (Database issue), D257-D260. (2014).
41. Keyes, R.M., Pejo, E., Katagiri, K., Huynh, K., Rudnitskaya, A., Stec, B., Stieglitz, K.A. Shape matters: Improving docking results by prior analysis of geometric attributes of binding sites. JSM Chem 4,1020-1024. (2016).
42. Soichet, B.K., Kuntz, I.D., Protein docking and complementarity. J. Mol. Biol. 221, 327-346. (1991).
43. Leiros, I., Mcsweeney, S., Hough, E.The Reaction Mechanism of Phospholipase D from Streptomyces Sp. Strain Pmf. Snapshots Along the Reaction Pathway Reveal a Pentacoordinate Reaction Intermediate and an Unexpected Final Product J.Mol.Biol. 339: 805 (2004).
44 Magotti, P.,Bauer, I.,Igarashi, M.,Babagoli, M.,Marotta, R.,Piomelli, D.,Garau, G. Structure of human N-acylphosphatidylethanolamine-hydrolyzing phospholipase D: regulation of fatty acid ethanolamide biosynthesis by bile acids. Structure 23: 598-604. (2015)
45. OGuin, T.H. et al. Phospholipase D facilitates efficient entry of influenza virus, allowing escape from inate immune inhibition. J. Biol. Chem. 289, 25405-25417 (2014).
46. Lapierre, J.M., Eathiraj, S., Vensel, D., Liu, Y., Bull, C.O., Cornell-Kennon, S., Iimura, S., Kelleher, E.W., Kizer, D.E., Koerner, S., Makhija, S., Matsuda, A., Moussa, M., Namdev, N., Savage, R.E., Szwaya, J., Volckova, E., Westlund, N., Wu, H., Schwartz, B. Discovery of 3-(3-(4-(1-Aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amine (ARQ 092): An Orally Bioavailable, Selective, and Potent Allosteric AKT Inhibitor. J.Med.Chem. 59: 6455-6469. (2016)
47. Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK Server: Interactive Docking Prediction of Protein-Protein Complexes and Symmetric Multimers. Bioinformatics 30(12): 1771-1773(2014).
48. https://www.chemaxon.com/download/marvin-suite/ (Last accessed 6/5/17)
49. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 25(13):1605-1612. (2004).
Table 1 Comparison of halopemide and FIPI inhibitory activities (IC50) toward hPLD1 and hPLD2
PLD1 PLD2
halopemide FIPI halopemide FIPI
Cellular 21 nM 1 nM 300 nM 44 nM
Biochemical 220 nM 9.5 nM 310 nM 17 nM
Figure Legend
Figure 1 Phospholipase D activity and generation of bioactive lipid PtdOH. A. The structure of phosphatidylcholine (PC) PLD catalyzed conversion to Phosphatidic acid (PtdOH). Note that PC is the preferred substrate for many PLD’s although they do cleave other glycerophospholipids. The cleavage site is the P-O bond adjacent to the choline head group (arrow) liberating PtdOH and free choline. Figure was drawn in ChemAxon’s Marvin Suite (48).
B: Schematic drawing of the different lipids generating or generated from PtdOH. PC, phosphatidylcholine; PA, phosphatidic acid; LPA, lyso-PA; DAG, diacylglycerol; CDP-DAG, cytidine diphosphate-DAG; PLA2, phospholipase A2; LPAAT, LPA acyltransferase; PAP, PA phosphatase; DGK, DAG kinase; CDS, CDP-DAG synthase; mito PLD, mitochondrial phospholipase D.
Figure 2 The evolution of PLD isozyme-specific and universal inhibitors A. Chemical drawingshowing the direct PLD inhibitor halopemide and a second generation direct inhibitor FIPI. Note the change in the position and attachment to a different functional group of the fluorine atom. Also note the omission of chlorine atom on second generation inhibitor FIPI.Figure was drawn in ChemAxon’s Marvin Suite (48).
B Chemical drawing showing the transformation from FIPI (5-fluoro-2 indolyl des-chlorohalopemide) to a potent hPLD1 specific inhibitor with more than 4-fold lower off-target activities. Note the change in functional group(s) after the amide bond. Figure was drawn in ChemAxon’s Marvin Suite (48).
C Chemical drawing of second generation halopemide-based inhibitors from a selection of potent PLD inhibitor analogues known as the “triazaspirone series” [1]. Note the slight change in the NPTSC (N-phenyl-triazaspirone congener) structure on the left with the S-methyl group on the linker on the right (NPTSC_S-methyl). This small change in the structure resulted in a 40-fold more potent PLD1 inhibitor with IC50 of NPTSC at 1,000 nM for hPLD1 and 110 nM for hPLD2 whereas NPTSC_S-methyl exhibited IC50 25 nM for hPLD1 and 140 nM for hPLD2. Essentially the PLD isoform selectivity was reversed [1]. Figure was drawn in ChemAxon’s Marvin Suite (48).
D Chemical drawing of third generation halopemide-based inhibitors from a selection of potent PLD inhibitor analogues known as the “triazaspirone series” [1]. Note the changes from ML298 to ML299: addition of the S-methyl group on the linker region and omission of both fluorine atoms and addition of bromine on the phenyl functional group to the right of the amide bond adjacent to the S-methyl group on the linker. These changes resulted in ML299 becoming ~3,000 fold more sensitive to hPLD1 than ML298 while still remaining a potent inhibitor of hPLD2. Figure was drawn in ChemAxon’s Marvin Suite (48).
Figure 3 The first universal PLD inhibitor Chemical drawing of the first universal phospholipase D inhibitor. Arrows indicate 6-OH deletion site the result of which renders the universal inhibitor devoid of selective oestrogen receptor modulator (SERM) activity.The universal PLD inhibor has been tested on at least 4 types of PLDs: hPLD1, hPLD2, PldA and NAPE (N-acyl phosphatidylethanolamine-specific phospholipase D)-PLD. Figure was drawn in ChemAxon’s Marvin Suite (48).
Figure 4 Docking of FIPI into PLDpmf active site A. The halepomide-based inhibitor FIPI (magenta) is docked into the PLDpmf active site (blue residues) of PDB 1V0Y [43]. Figure was rendered in Chimera [49]. B Surface map of PLDpmf active site area of PDB 1V09 [43] with residues colored by element and FIPI (magenta) docked across the active site pocket (blue residues). Note the presence of hydrophobic pocket directly “behind” the FIPI molecule. Also note the fluorine is pointing towards an acidic pocket (aspartic acid residues). Depending on the protonation of the residues in this pocket, docking observed suggests hydrogen bonding with fluorine atom could occur. Figure was rendered in Chimera [49]
Figure 5 Docking of ML299 into possible PLDpmf allosteric binding site. PLDpmf is shown (blue) with short chain phosphatidic acid product co-crystallized with the protein in the active site of PDB 1V0Y (43). The short chain lipid product is drawn as a sphere with conserved H170 shown in the active site. Directly below ML299 shown as a sphere is docked in a hydrophobic pocket. Conserved residue S167 is observed to be in close proximity to the bromine atom on the potent hPLD inhibitor. Figure was rendered in Chimera [49]
Figure 6 Docking of PLD universal inhibitor into the NAPE-PLD PDB 4QN9 [44] active site. A PLD universal inhibitor is shown in green and NAPE-PLD residues in gray. Note the universal inhibitor is observed in docking studies to block access to the PLD-NAPE active site.
B Surface map of NAPE-PLD 4QN9 [44] Surface rendered by element color. NAPE-PLD active site residues are grey, universal inhibitor green. Note the inhibitor is docked in close proximity to a hydrophobic pocket adjacent to the active site. Figure was rendered in Chimera [49]
Figure 7 Docking of PLDpmf PDB 1V0Y with AKT PDB code 5KCV [46] The PLDpmf PDB 1V0Y [43] is shown (magenta) with co-crystallized short chain phosphatidic acid (sphere colored by element) suggested to form a heterodimer with AKT PDB (blue) 5KCV [46] The allosteric inhibitor of AKT 3-(3-(4-(1-Aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amine (ARQ 092) (rendered as sphere colored by element) is far to the right of the suggested heterodimer interface of the two proteins observed in docking studies. Figure was rendered in Chimera [49].
Figure 1A
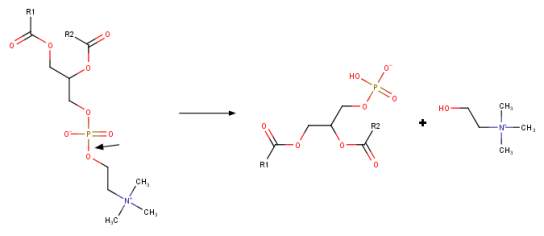
PLD
Phosphatidylcholine
Choline
Phosphatidic Acid
Figure 1B
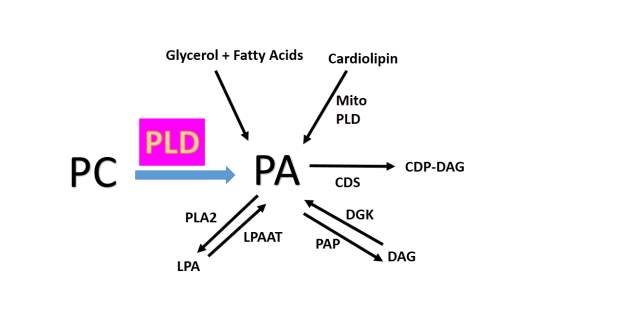
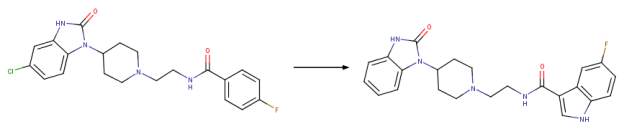 Figure 2A
Figure 2A
Halopemide
FIPI
Figure 2B
FIPI
VU0359595
Figure 2C
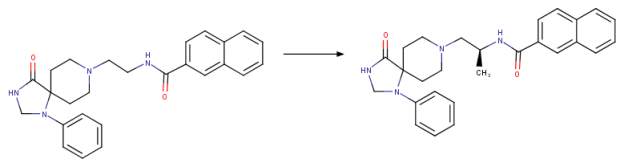
NPTSC
NPTSC_S-methyl
Figure 2D
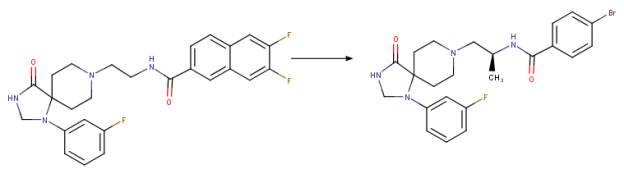
ML299
ML298
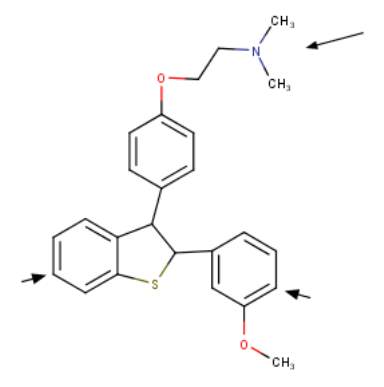
Figure 3
PLD Universal Inhibitor
Figure 4A
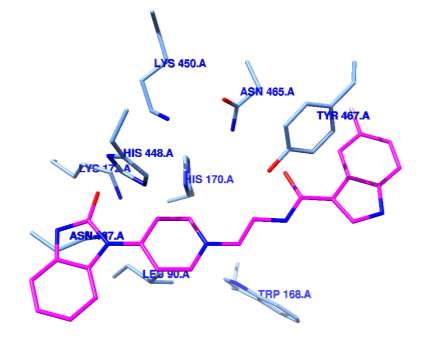
Figure 4B
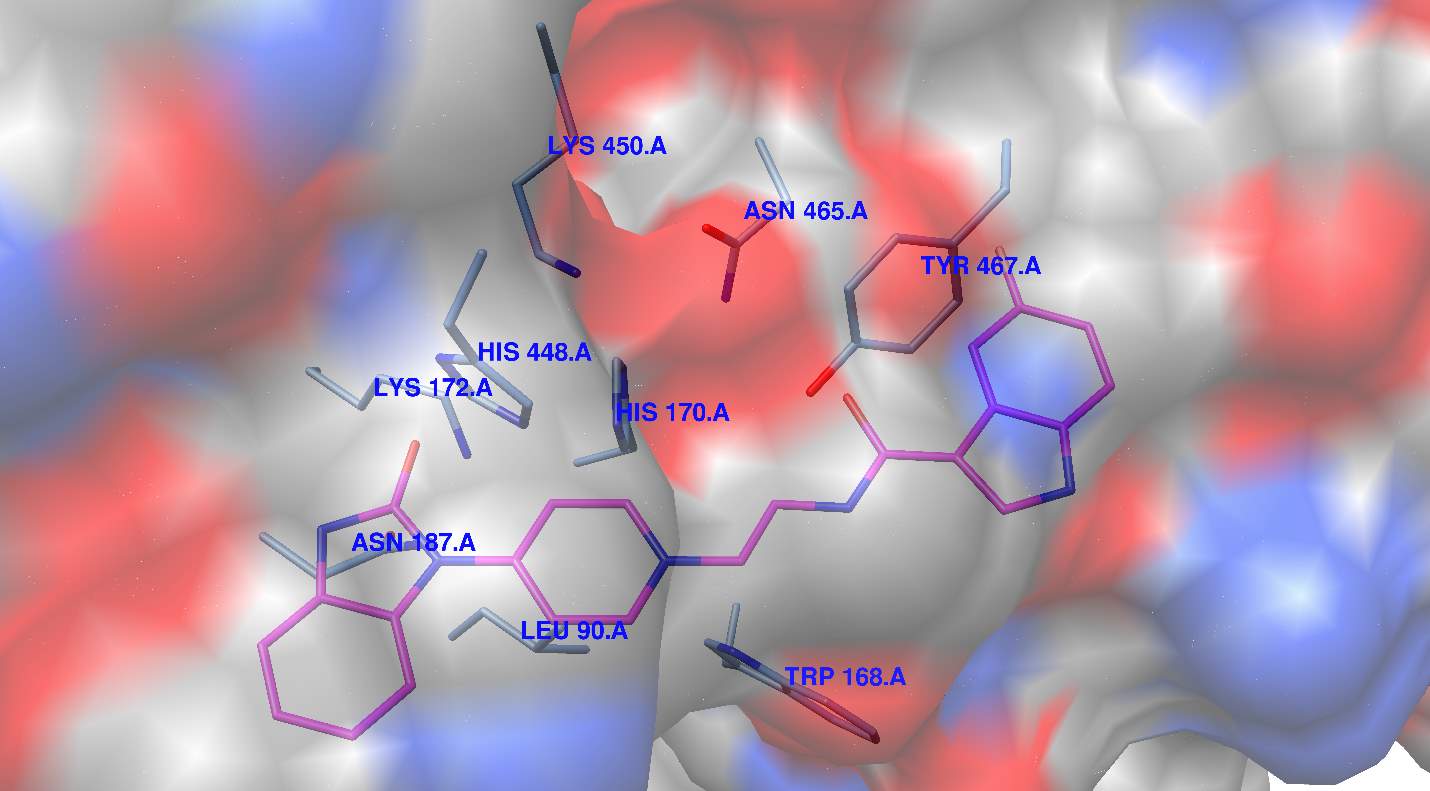
Figure 5
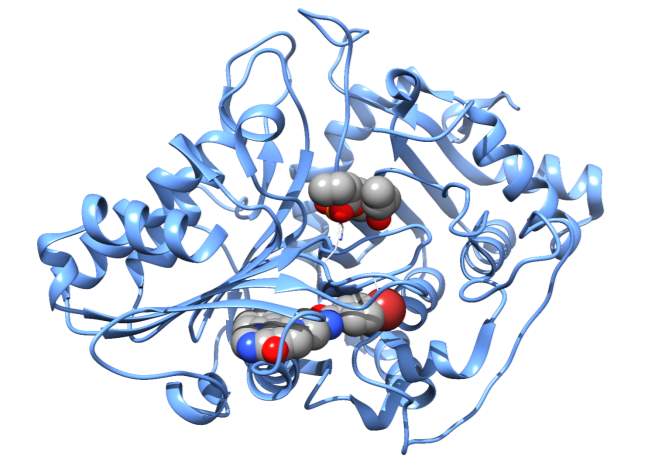
Figure 6A
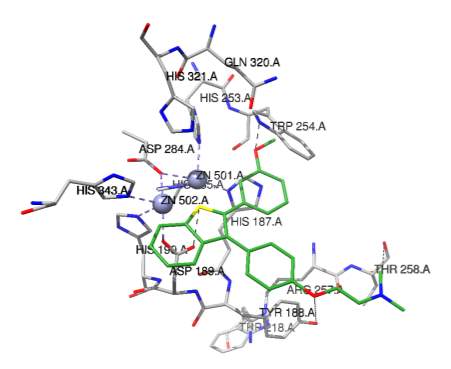
Figure 6B
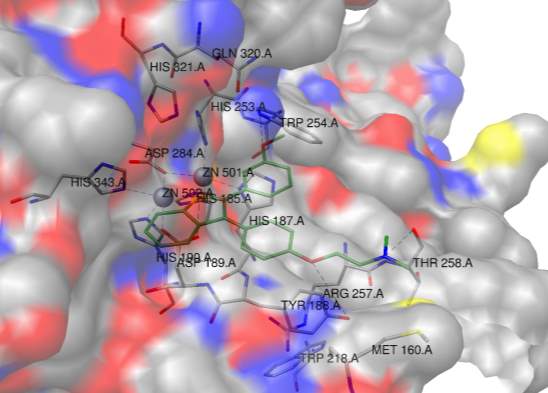
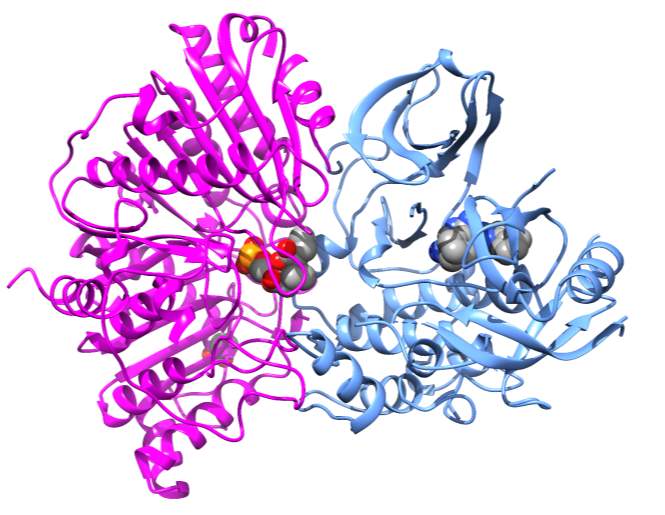
Figure 7
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biomedical Science"
Biomedical Science focuses on how cells, organs and systems function in the human body and underpins much of modern medicine. Biomedical Science applies parts of natural and/or formal sciences to help develop advances in healthcare.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: