Exploration of Catechol based Redox Non-Innocent Ligands in Redox Flow Batteries
Info: 6897 words (28 pages) Dissertation
Published: 10th Dec 2019
Tagged: Electronics
Contents:
Page
1. Energy Storage: 3.
2. Redox Flow Batteries (RFBs): 4.
2.1. RFB components: 4.
2.2. RFB electrochemistry: 6.
2.3. RFB advantages and disadvantages: 7.
2.4. Inorganic RFBs: 8.
2.5. Organic RFBs: 9.
3. Redox Non-Innocent Ligands: 10.
3.1. Catechols In Electrochemistry: 12.
3.2. Transition Metal Catechol Complexes: 13.
3.3. Functionalisation and Property Tuning of Catechol Derivatives: 13.
4. Characterisation of metal-ligand complexes for flow batteries: 14.
5. Summary and Project Objectives: 15.
6. References: 16.
1. Energy Storage:
In the last few years the UK Government has put growing emphasis on changing our energy paradigm from fossil fuels to clean renewable energy sources. With renewables only producing approx. 25% of our power demand the inherent instability of renewable energy sources is not currently an issue as the power produced by green methods serves only to reduce our use of fossil fuels.1 However, if we are to meet any serious green energy targets and eventually move past our dependence on fossil fuels we must first find a way to smooth out the immutable inconsistencies of photovoltaic solar and wind power which are as yet the most cost effective forms of renewable energy generation in the UK.2
If we are to meet the governments targets for renewable energy our related energy storage technologies must evolve and diversify to meet the demands of a growing infrastructure and market dynamics by becoming more energy dense, more cost effective, safer and environmentally friendly. Currently the most predominant form of energy storage around the world is pumped hydro, accounting for 130GW of stored power in 2016.3 While pumped hydro energy storage (PHES) has so far been a clear winner for its economy of scale, this form of power storage has several key drawbacks such as low energy density leading to increased tank height to increase gravitational potential energy or landscape reliance to increase volumetric capacity which can lead to environmental damage from damming.4
The most pressing issue with PHES is its power latency, requiring on the order of minutes to switch state between charging and discharging, while this might not sound like a long time this relegates the whole technology to secondary power management scenarios such as peak shaving. Clearly the efficacy of this technology has already reached its peak and to move forwards we must prove newer technologies that will become the more economically favourable option when considering construction costs, power latency and storage scalability.5 These are areas where electrochemical systems such as batteries, capacitors and fuel cells are already technologically dominant with regards to power latency and provide many distinct possibilities with respect to installation cost and capacity scaling in the primary energy management field.
2. Redox Flow Batteries (RFBs):
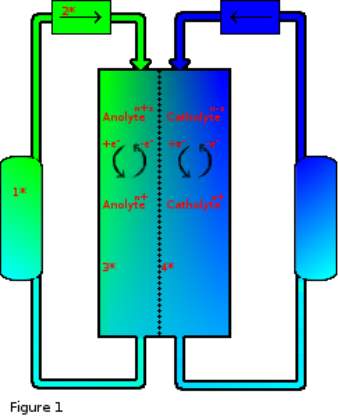 The concept of RFBs has been investigated since 19496 and while progress has been made7, 8 the technology has been largely bypassed by more energy dense or otherwise convenient forms of energy storage. Li-ion batteries for example excel in scenarios where portability (power/weight) is favoured such as in mobile phones and electric vehicles. However, Li-ion batteries suffer heavy compromise by way of battery longevity and even safety.9 In situations where portability is not an issue however RFBs have also been overlooked for more mechanical methods such as pumped hydro energy storage, compressed air or heat storage, and while these forms of energy storage are scalable and effective they each have downsides such as environmental disruption, power latency, energy loss, even safety concerns in some cases. All of which turn out to be key strengths of a fully developed RFB system.
The concept of RFBs has been investigated since 19496 and while progress has been made7, 8 the technology has been largely bypassed by more energy dense or otherwise convenient forms of energy storage. Li-ion batteries for example excel in scenarios where portability (power/weight) is favoured such as in mobile phones and electric vehicles. However, Li-ion batteries suffer heavy compromise by way of battery longevity and even safety.9 In situations where portability is not an issue however RFBs have also been overlooked for more mechanical methods such as pumped hydro energy storage, compressed air or heat storage, and while these forms of energy storage are scalable and effective they each have downsides such as environmental disruption, power latency, energy loss, even safety concerns in some cases. All of which turn out to be key strengths of a fully developed RFB system.
2.1. RFB components:
Figure 1 key:
- Electrolyte tank.
- Pump.
- Electrode surface.
- Semi-permiable membrane.
A typical RFB is more mechanically complicated than most other battery designs which usually have no moving parts. RFBs on the other hand rely on pumps to cycle electrolytes from tanks into their electrochemical cells, with most designs as shown using two tanks, one each to separate anolyte from catholyte. Some electrolyte solutions are corrosive due to high aqueous acid or base content, so tanks in these cases are typically chemically resistant polymers. Some non-aqueous designs utilise solvents that may not be compatible with certain polymer types so there is variability among polymer choice. Tanks can also be any size, resulting in capacity independent characteristics of RFBs.6
Pump choice is also a variable to consider although choices tend to focus on either centrifugal or peristaltic designs, as it is important to both separate the pump electrical components from electrolytes and maintain consistent flow dynamics, both types of pump can fulfil these requirements however they are not the only options. The energy requirement of the pump is also a source of inefficiency within the battery and typically pumps are run at the minimum required speed to conserve power and maintain steady state at electrodes with the required charge/discharge rate.6
Electrodes are one area with a large degree of available materials and many reasons for specific material choice. The electrodes, also referred to as current collectors, can be any conductive material however for both cost and chemical stability reasons graphite, amorphous and other forms of carbon are extremely common. These carbon-based electrodes are sometimes doped with various elements to improve electrode kinetics however this is not a necessity.10 Metal electrodes are also possible but can be susceptible to corrosion and impurity poisoning, they are also more difficult to provide in a form that maximises surface area. Carbon based electrodes are typically low cost and high surface area so are a clear choice industrially however, it has been found that sometimes the use of carbon electrodes can result in irreversible electrochemical reactions.11
Membranes are an important feature of most cell designs and while not strictly necessary to RFB design12 provide an extremely important function of separating anolyte from catholyte to prevent auto-discharge and electrolyte poisoning. The semi-permeable membrane also allows the passage of charge balancing ions that would otherwise accumulate and prevent further electrochemical discharge. The choice of which type of membrane to use is governed mostly by the solvent choice but also by counter-ion conductivity, membranes in aqueous solution are typically Nafion as this commercially available material has one of the highest proton conductivities available however for other solvents and where protons are not the counter-ion other choices are available with typically much reduced conductivities.13 Membranes are a large source of internal resistance within a flow cell and attempts have been made to bypass this limitation such as laminar flow and immiscible fluid dynamics.14
2.2. RFB electrochemistry:
The electrochemical profile of a RFB, its specific power (the amount of energy that can be instantaneously be released per electrode surface area), its energy density (the total amount of energy held by the system per weight), and its discharge voltage are all determined by the specific chemistries of the battery.
Solvent choice is a key factor as any redox couples that exceed the breakdown voltage of the solvent will incur significant efficiency losses during charging due to solvent electrolysis, this limits aqueous systems to 1.23V. Inorganic solvent choice is usually determined by chemical compatibility and the molecular polarizability15 which effects two considerations when designing a RFB, the electric double layer length which has a direct effect on electrode kinetics hence specific power and the electrolyte solubility by screening the charge of dissolved ions potentially allowing more to dissolve therefore increasing energy density.
Redox mediators are also an important class of additives that can significantly improve electrode kinetics and even battery longevity when unintended side reactions cause unintended deposition of solid electrolytes during discharge. Redox mediators are activated by providing an overvoltage while charging and act as an electron hole transfer agent and when solid by-products are present can be used to regenerate active electrolyte.16
Flowing electrolytes are the main ingredient of all RFBs however some systems exist by combining a flow half-cell with mixed phase redox couples such as lithium-TEMPO17 or sulphur-oxygen,18 this type of system is referred to as a hybrid RFB. While the solvent of the system may limit the voltage because of its unwanted electrolysis the resulting voltage of the battery is defined by the total energy gap between the states of both anolyte and catholyte however voltage is not the only factor controlling the energy density of a resulting system. Charge stoichiometry is also extremely important as each reversible electron transfer process can be utilised to produce electricity. More specific electrolyte chemistry will be explored in the following sections.
2.3. RFB advantages and disadvantages:
RFBs have many advantages over more common counterparts:
- Highly scalable storage, limited only by tank size.
- Power / capacity independence, unlike other kinds of battery that typically scale output directly with capacity, the power of a RFB is decided by the surface area.
- Physical separation of electrolytes means RFB cells can’t auto-discharge or deteriorate over time.
- Even under short-circuit and other failure conditions the battery remains relatively safe with electrolytes acting to remove heat and hence not cause fire or other cell damage.19
- Cells typically function at ambient temperature and pressure reducing complexity and maintenance costs.
- The potential ability to use the same electrolyte for both oxidised and reduced sides of the battery protects the battery from contamination over time and thus electrolyte crossover results only in power lost and not poisoned electrolytes with reduced efficiency or complicated maintenance processes.
- Not affected by state of charge or depth of cycle, unlike other kinds of battery where maintaining full or empty states can cause electrode damage due to side reactions or dendrite formation.
- RFB electrodes do not change with reaction progress and hence stay in a stable state and through proper electrolyte development side reactions are ideally prevented that would prevent full capacity use.20
Unfortunately, the current state of RFB technology has several key disadvantages including relatively low energy density, around 10-50 Wh/Kg although this low energy density is representative of older systems further examples will be given below.21 Low specific power, which while not an issue for large scale static applications as variable numbers of cell stacks can be used to control voltage and as much surface area as needed can be used for current, however this limitation effectively preclude its use in electric vehicles and mobile applications.6
These comparisons are typically made against a modern Li-Po design which can hold on the scale of 100-265 Wh/Kg,20 and as a solid-state battery with no moving parts the specific power isn’t limited by flow rates and semi-permeable membrane resistances etc. Other work with organic electrolytes have reported potential energy densities of up to 188 Wh/L with a Li/TEMPO hybrid system.22 This and other developments are significant improvements but still sub-par when considering the system(s) as a whole when compared with other available energy storage systems and their use cases.
2.4. Inorganic RFBs:
Of the inorganic class of electrolyte materials aqueous vanadium has been researched most thoroughly showing promise for several reasons such as reasonable electrochemical potential of 1.45 V, high columbic efficiency as the water is not electrolysed until above 90% capacity, excellent reaction reversibility resulting in better RFB longevity. However, this system is hampered by the disadvantages discussed above, with low energy density, and current densities of approx. 50 mA cm-2 and energy density of 25+ Wh/Kg .21 Other redox couples have been explored within the scope of inorganic molecules, achieving good cell voltages as high as 3.4 V with chromium(acac)3 in acetonitrile however extremely low (20%) efficiency,23 with cobalt chloride in acetonitrile at 2.1 V with decent (85%) efficiency.24 The reaction kinetics of typical metals also limits the charge/discharge characteristics significantly pushing research into catalysis of the electrode reactions.25, 26 The solubility of metal centres also depends heavily on how acidic the solution is, this heavily reduces the solubility of metal species in organic solvents, increasing costs with electrolyte complexity and raising risks associated with strongly acidic solutions in case of a failure condition.24
These inefficiencies are typically caused by solvent breakdown such as water being hydrolysed to H2 and O2 which has pushed inorganic experiments towards organic solvents such as acetonitrile, DMSO, DMS, etc.27 Organic solvents are often employed to maintain higher cell voltages without electrolytic losses however the use of these solvents typically also reduces the solution conductivity which is largely dependent on active species mobility and the polarizability of the solvent, and hence specific power of the cell is typically reduced with organic solvents.28 The investigation of supercritical solvents has been approached and shows some potentially appealing features such as gas like diffusivity however the difficulty working with these materials has restricted their investigation significantly.29
Aside from metal centres other inorganic redox couples have been investigated including polysulphide/iodine achieving an impressive 43.1 Wh/L and specific power of 37 mA cm-2 when considering greatly reduced cost of materials, approx. half the cost per capacity of a comparable vanadium based application.30 This system of sulphur and halogen did not require the use of additional acid to increase solubility as the sulphide and halogen were already quite soluble in aqueous mediums.
2.5. Organic RFBs:
Organic compounds show more promise as suitable charge carriers for RFBs. Once developed they are cheap to make as they do not require potentially rare transition metals and their properties can be honed to specific voltages and charge densities within the potential window of the supporting solvent.22, 31 This combination of factors shows the strength of organic compounds such as quinones (2-vinyl-anthraquinone) to hold charge(s) without compromising molecule stability and have been shown to demonstrate orders of magnitude higher electrode kinetics than metallic systems without catalysis.27 Although in the cited case the organic species is applied to a polymer backbone immobilizing it for use as a solid, conversely RFBs work more effectively with increasing solubility of redox couples.22, 32
Other work has attempted to use inspiration from biological charge carriers and has explored the use of flavonoids as the electroactive specieswith energy densities around 284 Wh/Kg with a riboflavin/Li hybrid system.33 It is also worth noting that any biological molecule can be produced efficiently via biological methods then derivatised, greatly reducing costs incurred by stepwise synthesis from start to finish. The electrochemistry of readily available dye compounds such as Indigo, alizarin and quinizarin has also investigated showing results on par with vanadium RFBs in much milder solution conditions with minimal functionalization to increase aqueous solubility.32 The conjugated delocalised nature of compounds such as alizarin, quinizarin and quinones have been shown to facilitate rapid (proton coupled) electron movement in aqueous environments extremely well, showing rate constants 2-3 orders of magnitude higher than that shown in vanadium based systems in some cases.34
The redox couples described above have all had relatively high energy densities when contrasted to vanadium systems because each has typically undergone two electron transfer processes. The two-electron transfer effectively doubles the energy density if other variable such as cell voltage, faradic efficiency and concentration remain constant as shown in successful application of viologen providing decent energy density of 133 Wh/L and improved specific power of 94 mA cm-2.35 Unfortunately while the kinetics is much better than that of metal based systems the current density is still hampered by semi-permeable membrane dynamics.8
Unfortunately, organic redox couples are often capable of undergoing side reactions when under conditions that are unfavourable to their stability. This is particularly apparent with radical species and must be taken into consideration when designing the electrolyte system. Molecules that can act as conjugate acids when holding extra electrons typically gain a stability bonus however this is typically limited to aqueous systems. Radical side reactions can also be avoided by the utilisation of two electron transfer processes and or steric protecting groups.36
Where the same electrolyte is used for both reduction and oxidation, because the molecule can exist in three states oxidised, neutral, and reduced, several benefits are observed such as no electrolyte poisoning by cross contamination. This raises the possibility of avoiding semi-permeable membranes entirely by the careful application of laminar flow dynamics or other techniques,12 potentially significantly reducing cell resistance and allowing much higher current densities. The use of a single electrolyte, while costing some efficiency via contamination also entirely depreciates the need for costly maintenance steps such as electrolyte purification.
3. Redox Non-Innocent Ligands:
A redox non-innocent ligand (NIL) is one which shows the ability to exist in more than one oxidation state or an oxidation state not accurately described by the typical oxidation states of its constituent atoms.37 A common example of a NIL is catechol/1,2-benzoquinone that when bonded to a metal centre the arrangement of atoms is identical only the bonding structure can change allowing reversible and rapid oxidation or reduction between forms. The function of NILs while only more recently investigated by humanity has long been exploited in biological catalysis, by allowing complexes to have oxidation states that would be otherwise disfavoured by the metal centre.38 This has so far found use in tuning the properties of cheaper catalysts to mimic that of more expensive metal centres, however the redox potentials achieved by metal-ligand tuning could also be used to improve the energy density in RFBs.
This biological use of NILs can be replicated and/or re-tasked to our specific use cases by application of suitable functional groups to “tune” the redox potential and solubility of the resulting complex.39 The specific properties of azoles-pyridine type ligands have been explored and show that the electrolyte potential window can be expanded significantly, from 1.07 to 1.91 V with the explored Co(NIL) system, by decreasing the degree of σ donation into and increasing π acceptance by the ligand from the metal centre.11 This method of increasing battery capacity is most effective in non-aqueous systems, as they are not limited by the narrow potential window of water (1.23 V), however when pushed too far challenges can arise in synthesis of the complex and electrode reaction reversibility issues can also arise.
However, this is not the only way NILs can be used to our advantage, as a species that can hold or lose extra electrons this class of ligands can also theoretically act as a charge reservoir.40 The charge capacity increase of the complex is possible because the charge of the NIL and the metal centre form a kind of equilibria with rapid and free flow of electron density within the complex.37
When considering the possible combination of factors looked at above it is apparent that within the scope of possible RFB systems there are several different dimensions of variability that have each been investigated individually and incrementally improved upon. These bottom up variables such as solvent choice, electrolyte concentration, reaction kinetics, metal centre, ligand chemistry, even mechanical cell properties and membrane choice when compared with the resulting top down battery properties of cell voltage, electron stoichiometry, faradic efficiency, and cell resistance together hint at the possibility of greatly improved battery performance across all key weaknesses of previous technologies. This hints at possible combinations of redox flow battery technologies that have far surpass the key weaknesses of older RFB technologies and potentially even surpass current battery systems on energy density. However, this challenge of finding suitable ligands to maximise both potential gap and electron number is a delicate balance of stability and instability between multiple electronic states within the same complex and may prove to be analogously difficult to therapeutic drug design.
3.1. Catechols In Electrochemistry:
The electrical properties of catechol are being investigated not only in batteries where the long-term storage of energy is the fundamental goal, but also in electrochemical capacitors where power needs to be released extremely rapidly. Catechols have been shown to demonstrate a high rate of reaction at the electrode surface and high reversibility lasting upwards of 10k cycles.41 In capacitors and the same for RFBs the coupled 2-electron 2-proton transfer is analogous to the reaction shared by quinones,31 with the extra electron density being distributed among the newly formed O-H bonds thus providing extra stability to the molecule. Or in the case of the reverse reaction the [H+] counterion is released preventing electron deficiency. The molecule also has relative stability in the radical state because of electron delocalisation between the ring and oxygen functional groups.
Figure 2 shows 1) 1,2-benzoquinone 2) semiquinone radical and 3) catechol reversible redox conversions.
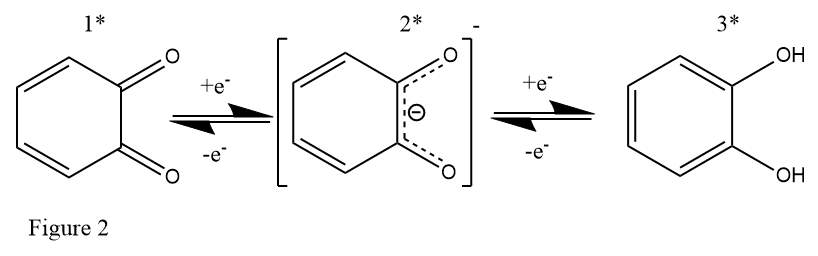
Electrochemical oxidation of flavonoids containing the catechol moiety has also been reported42 however the results for use in RFBs are less promising for this class of molecules as the oxidised flavonoid is unstable with subsequent rearrangement and reduced aromaticity with further susceptibility to oxidation by oxygen and loss of the catechol moiety. This is possible because of the more stable and less asymmetrical quinone moiety being produced and the proximity of nearby groups to rearrange into the more favourable state. Potential structural rearrangements of derivatised catechols should also be avoided as to generate more stable electrolysis products for the long-term storage of energy as is the purpose of a battery.
 3.2. Transition Metal Catechol Complexes:
3.2. Transition Metal Catechol Complexes:
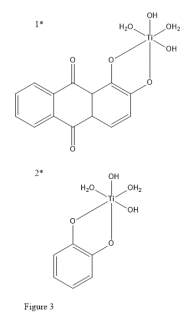
When comparing the electronic properties of catechol to other ligands (alizarin) it has been shown by Duncan et. al.43 that in catechol the ground state for a neutral Ti(catechol)(H2O)2(OH)2 (Figure 3, 2) electron density is preferentially stored on the catechol, unlike the alizarin analogue Ti(alizarin)(H2O)2(OH)2 (Figure 3, 1) where the Highest Occupied Molecular Orbital (HOMO) is primarily located on the Ti centre. This implies that when multiple catechols are bound to titanium, and potentially other metal centres, that as indicated by their HOMO, excess electron density is directed into the catechol and is not attempting to concentrate in the metal centre which would reduce stability. Their work also indicates a possible route to increase the voltage gap of resulting complexes via substituent tuning as indicated by increasing π back-bonding character.11
Catechols can complex with various metal centres with varying degrees of bond strength due to orbital mechanics of the metal. This is because of varying degrees of σ and π character between the components of the complex. In the study the resulting bonding structures also show strong preference for the number of ligands attached to each centre. This is also indicative of the bonding strength of the ligand.44 While this study did not perform electrochemical analysis of the complexes the resulting binding strength differences show a strong case for increased complex stability as the energy required to separate the ligand from its metal centre is more difficult to surpass.
3.3. Functionalisation and Property Tuning of Catechol Derivatives:
When deciding how best to functionalise a ligand in order to increase the aqueous solubility of a resulting complex several simple methods, one of these being sulfonation with to prevent precipitation,22 this has proven effective with organic molecules however SO3-2 is a large group and would reduce the mass efficacy of the electrolyte. Sulphonation is not the only method however and other techniques such as providing hydrogen-bonding sites or dipoles32 and perhaps reducing the exposure of aromatic areas of the complex to the solvent could be explored. As much of the surface of the complex is organic in nature this may be an appropriate route to take.
One of the most important factors to increase power density is by increasing the range of charges each complex can hold, this is shown on the ligand side with simple catechols by the reversible oxidation between catechol and 1,2-benzoquinone. This ability to hold charge is provided by ease of bond rearrangement without changing the molecular structure,41 and can in theory be expanded upon by introduction of suitable groups such as carbonyls that can readily be reduced into alcohols or heteroatoms such as nitrogen that can donate an electron density into its aromatic ring. As with other areas of organic chemistry there are many different approaches that can result in functional groups that act as reversibly charge donors and acceptors. The other side to this is the preferred oxidation states of the metal centre, and while the use of non-innocent ligands allows the complex to more easily cross barriers to differing electronic states40 the metal centre employed typically still exists in its more favourable oxidation states, thus selection of the metal centre should not be oblivious to the range of charge afforded by the transition metal.
The potential difference is another key factor in the performance of a battery and is proportional to the amount of energy stored. This can be modified by changing the bonding character of the ligands, such that increasing the π accepting character of the ligand appears to increase the difference between energy levels.11 This π accepting character can be increased by application of electron withdrawing groups such as halides, carboxylic acids, and cyano groups. The potential difference is also changed by the choice of metal centre and the effect of varying the π accepting character will surely be modified by the orbital characteristics of the metal.
The stability of simple catechols is less than ideal with many research papers dedicated to organometallic catalytic cleavage,45, 46 as we are investigating catechol-based complexes this is an important factor to take into consideration. Typical catalytic breakdowns occur with complexes designed to mimic biological enzyme complexes in the presence of oxygen. Maintaining an oxygen free environment may therefore also be of utmost importance during the project.
4. Characterisation of metal-ligand complexes for flow batteries:
To determine the bonding structure of a ligand-metal complex IR spectroscopy is a good choice as the peak wavenumbers can provide information on bond type and strength via up/downfield shifting.39 The application of UV-Vis spectroscopy can also determine low energy transitions that might potentially reveal molecular orbitals that can store a charge within the complex. 1H and 13C NMR is always a strong choice for use in determining organic structures and will be invaluable in the analysis and preparation of any ligands to be investigated. Various NMR experiments such as 2D COSY can be used when typical proton-proton coupling is not sufficient to discern chemical structure.47
The electrochemistry of a potential redox couple is commonly determined by cyclic voltammogrammy, a technique that shows redox transitions and required voltages to drive them. This technique also shows irreversible electrode reactions when a potential electrolyte species is driven by sufficient overpotential to cause unwanted reactions.39
Real time IR and UV/Vis spectra can also be utilised during cell cycling to determine short lived intermediates and side reaction pathways in the event of unexpected chemical reactions in order to prevent or exploit these effects.47
5. Summary and Project Objectives:
From considering the available material, it is clear there are many potential routes of investigation within the scope of catechol complex based redox flow batteries. I think the most pertinent factors to addressing the low energy density of RFBs can be approached from multiple angles even simultaneously, by first finding ligand variants that remain stable across several redox states, hopefully approaching or exceeding ±3 on each ligand, while also maintaining a strong bond to the metal centre so that the produced electrochemical characteristics remain consistent. The second factor to consider when designing the ligands to be assessed is the effect of various substituent groups on the voltage gap between oxidised and reduced species.
In this project going forwards I think it would be best to focus on aqueous systems despite limiting the potential window to 1.23V or less as to take advantage of the increased conductivity and potentially increased solubility afforded by a more polar proton conducting solvent. It would also be useful to investigate the specific power characteristics of the set of complexes.
While the bonding strength of various metal centres with catecholates has been investigated44 I think it would still be worthwhile to assess multiple transition metal complexes such as titanium, iron and manganese to assess if they change the effective stability of the complex across their range of redox states and to see if any compromise can be made between resulting stability and voltage. As the set of ligands remains the same this should result in a greater picture of possible variations without adding too much to the complexity of the experimentation.
The resulting combinations of ligand and metal complexes should each be fully assessed first to identify that they are the intended products via suitable spectroscopic methods and that their physical characteristics such as solubility and any possible sensitivities to oxygen etc are known, then electrochemical techniques such as cyclic voltammogrammy to determine properties such as voltage gap, electron transfer reversibility and kinetics.
6. References:
1. E. I.S, 2016.
2. P. Alotto, M. Guarnieri and F. Moro, Renewable and Sustainable Energy Reviews, 2014, 29, 325-335.
3. E. Barbour, I. A. G. Wilson, J. Radcliffe, Y. Ding and Y. Li, Renewable and Sustainable Energy Reviews, 2016, 61, 421-432.
4. S. Rehman, L. M. Al-Hadhrami and M. M. Alam, Renewable and Sustainable Energy Reviews, 2015, 44, 586-598.
5. M. A. Escalante Soberanis, T. Mithrush, A. Bassam and W. Mérida, Renewable Energy, 2018, 115, 547-557.
6. L. F. Arenas, C. Ponce de León and F. C. Walsh, Journal of Energy Storage, 2017, 11, 119-153.
7. L. S. J. Giner, K. Cahill, Journal, 1976.
8. B. Hu, C. Seefeldt, C. DeBruler and T. L. Liu, Journal of Materials Chemistry A, 2017, 5, 22137-22145.
9. P. J. Mankowski, J. Kanevsky, P. Bakirtzian and S. Cugno, Burns, 2016, 42, e61-e64.
10. J. Kim, H. Lim, J.-Y. Jyoung, E.-S. Lee, J. S. Yi and D. Lee, Electrochimica Acta, 2017, 245, 724-733.
11. C. G. Armstrong and K. E. Toghill, Journal of Power Sources, 2017, 349, 121-129.
12. P. Navalpotro, J. Palma, M. Anderson and R. Marcilla, Angewandte Chemie International Edition, 2017, 56, 12460-12465.
13. G.-J. Hwang, S.-W. Kim, D.-M. In, D.-Y. Lee and C.-H. Ryu, Journal of Industrial and Engineering Chemistry, DOI: https://doi.org/10.1016/j.jiec.2017.11.023.
14. M. O. Bamgbopa, S. Almheiri and H. Sun, Renewable and Sustainable Energy Reviews, 2017, 70, 506-518.
15. M. O. Bamgbopa, N. Pour, Y. Shao-Horn and S. Almheiri, Electrochimica Acta, 2017, 223, 115-123.
16. Y. X. Ren, T. S. Zhao, M. Liu, Y. K. Zeng and H. R. Jiang, Journal of Power Sources, 2017, 361, 203-210.
17. P. Leung, A. A. Shah, L. Sanz, C. Flox, J. R. Morante, Q. Xu, M. R. Mohamed, C. Ponce de León and F. C. Walsh, Journal of Power Sources, 2017, 360, 243-283.
18. Z. Li, M. S. Pan, L. Su, P.-C. Tsai, A. F. Badel, J. M. Valle, S. L. Eiler, K. Xiang, F. R. Brushett and Y.-M. Chiang, Joule, 2017, 1, 306-327.
19. A. H. Whitehead, T. J. Rabbow, M. Trampert and P. Pokorny, Journal of Power Sources, 2017, 351, 1-7.
20. S. Jiao, J. Zheng, Q. Li, X. Li, M. H. Engelhard, R. Cao, J.-G. Zhang and W. Xu, Joule, 2017, DOI: https://doi.org/10.1016/j.joule.2017.10.007.
21. M. Rychcik and M. Skyllas-Kazacos, Journal of Power Sources, 1988, 22, 59-67.
22. Y. Ding, C. Zhang, L. Zhang, Y. Zhou and G. Yu, Chemical Society Reviews, 2017, DOI: 10.1039/C7CS00569E.
23. Q. Liu, A. A. Shinkle, Y. Li, C. W. Monroe, L. T. Thompson and A. E. S. Sleightholme, Electrochemistry Communications, 2010, 12, 1634-1637.
24. A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. Liu, Journal of Applied Electrochemistry, 2011, 41, 1137.
25. M. V. Holland-Cunz, J. Friedl and U. Stimming, Journal of Electroanalytical Chemistry, 2017, DOI: https://doi.org/10.1016/j.jelechem.2017.10.061.
26. L. Wei, T. S. Zhao, L. Zeng, Y. K. Zeng and H. R. Jiang, Journal of Power Sources, 2017, 341, 318-326.
27. G. L. Soloveichik, Chemical Reviews, 2015, 115, 11533-11558.
28. C. N. Sun, M. M. Mench and T. A. Zawodzinski, Electrochimica Acta, 2017, 237, 199-206.
29. K. E. Toghill, M. A. Méndez and P. Voyame, Electrochemistry Communications, 2014, 44, 27-30.
30. Z. Li, G. Weng, Q. Zou, G. Cong and Y.-C. Lu, Nano Energy, 2016, 30, 283-292.
31. W. Choi, D. Harada, K. Oyaizu and H. Nishide, Journal of the American Chemical Society, 2011, 133, 19839-19843.
32. J. Carretero-Gonzalez, E. Castillo-Martinez and M. Armand, Energy & Environmental Science, 2016, 9, 3521-3530.
33. M. Lee, J. Hong, D.-H. Seo, D. H. Nam, K. T. Nam, K. Kang and C. B. Park, Angewandte Chemie International Edition, 2013, 52, 8322-8328.
34. B. Yang, L. Hoober-Burkhardt, F. Wang, G. K. Surya Prakash and S. R. Narayanan, Journal of The Electrochemical Society, 2014, 161, A1371-A1380.
35. C. DeBruler, B. Hu, J. Moss, X. Liu, J. Luo, Y. Sun and T. L. Liu, Chem, 2017, DOI: https://doi.org/10.1016/j.chempr.2017.11.001.
36. H.-s. Kim, K.-J. Lee, Y.-K. Han, J. H. Ryu and S. M. Oh, Journal of Power Sources, 2017, 348, 264-269.
37. J. L. Boyer, J. Rochford, M.-K. Tsai, J. T. Muckerman and E. Fujita, Coordination Chemistry Reviews, 2010, 254, 309-330.
38. W. Kaim and B. Schwederski, Coordination Chemistry Reviews, 2010, 254, 1580-1588.
39. E. Safaei, H. Bahrami, A. Wojtczak, S. Alavi and Z. Jagličić, Polyhedron, 2017, 122, 219-227.
40. V. Lyaskovskyy and B. de Bruin, ACS Catalysis, 2012, 2, 270-279.
41. J. Choi, M. Yang and S.-K. Kim, Applied Surface Science, 2017, 422, 316-320.
42. H. P. Hendrickson, A. D. Kaufman and C. E. Lunte, Journal of Pharmaceutical and Biomedical Analysis, 1994, 12, 325-334.
43. W. R. Duncan and O. V. Prezhdo, The Journal of Physical Chemistry B, 2005, 109, 365-373.
44. B. P. Lee, A. Narkar and R. Wilharm, Sensors and Actuators B: Chemical, 2016, 227, 248-254.
45. M. Garai, D. Dey, H. R. Yadav, A. R. Choudhury, N. Kole and B. Biswas, Polyhedron, 2017, 129, 114-122.
46. E. Safaei, N. Naghdi, Z. Jagličić, A. Pevec and Y.-I. Lee, Polyhedron, 2017, 122, 116-123.
47. T. dos Santos Francisco, D. C. de Oliveira Cruz, A. A. Batista, A. G. Ferreira, J. Ellena, Í. de S. Moreira, E. H. S. Sousa, I. M. M. de Carvalho, E. Longhinotti and I. C. N. Diógenes, Polyhedron, 2012, 31, 104-109.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Electronics"
Electronics regards the science and technology involved in the development of electrical circuits and electronic devices and equipment that use them.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: