Activation of Various Signalling Pathways in Response to Treating Mouse Macrophage Cells with Lipopolysaccharides (LPS)
Info: 8152 words (33 pages) Dissertation
Published: 24th Feb 2022
Tagged: CancerBiomedical Science
Abstract
Liposaccharides (LPS) are the major components of the outer membrane that present in almost all Gram-negative bacteria. They play a significant role when it comes to activation of signalling pathways in mouse macrophage cells. The mouse macrophage cells line RAW264.7 was allowed to grow as two identical cultures. For the SILAC method, one of the cultures was labelled as ‘heavy’ with arginine and lysine and the other culture was used as control and it was labelled as ‘light’. The heavy labelled culture was treated with 100 units per ml of LPS for 30 minutes. Protein extraction, trypsin digestion and phosphopeptide enrichments were performed using equal amount of heavy and light cell cultures. LQT/Orbitrap Velos system was used to analyse the extracted phosphopeptide samples and generated data was analysed using Maxquant method. By using the Maxquant method, 41 significant upregulated peptides were identified, and they were used for KEGG pathway analysis. The uniprot accession of the significant upregulated peptides were used to identify the enriched KEGG signalling pathways.
The KEGG enrichment analysis suggested that there were 10 signalling pathways were activated in response to treating cells with LPS. The suggested signalling pathways were osteoclast differentiation, progesterone-mediated oocyte maturation, MTOR, MAPK, VEGF, Fc epsilon RI, prostate cancer, Toll-like receptor, Chagas disease (American trypanosomiasis) and cancer signalling pathways. However, further analysis proved that osteoclast differentiation, MAPK and Toll-like receptor signalling pathways were activated in response to treating mouse cells with LPS.
Introduction
Bacteria cell surfaces are decorated with various types of glycoconjugates that play critical roles in the interactions between bacteria and the environment [(Han et al., 2011)]. For example, the outer membrane of Gram-negative bacteria is mainly made up of lipopolysaccharides (LPS) that play critical roles in bacterial cell physiology and pathogenicity.
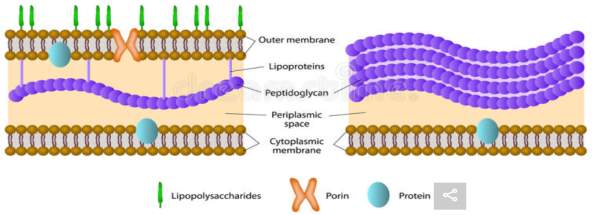
Figure 1: Gram-negative bacterium membrane structure (left) and Gram-positive bacterium cell membrane structure (right). Lipopolysaccharides (LPS) can only be found in the outer membrane of Gram-negative bacteria (shown in green above). LPS is a target for recognition by host antibodies because it is attached to the outer membrane of the bacterium cell. (https://www.dreamstime.com/stock-illustration-gram-positive-gram-negative-bacteria-difference-bacterial-image45337024) [2]
LPS is made up of three main structural components, a hydrophobic lipid section which is responsible for the toxic properties of the LPS molecule and secures the LPS structure in the outer membrane (lipid A), a hydrophilic core polysaccharide chain (inner and outer core region) and a repeating hydrophilic O-antigenic oligosaccharide side chain (O polysaccharide) that is distinctive to the bacterial serotype (Galanos, Lüderitz and Westphal, 2018) [3].
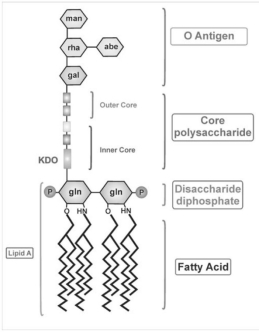
Figure 2: The general structure of lipopolysaccharides that attach to the outer membrane of the Gram-negative bacteria. LPS are made up of three distinct regions. Lipid A is made up of a β-glucosamine-(1→6)-glucosamine-1-phosphate base with fatty acid esters attached to both carbohydrates. The inner core made up of between 1 and 4 molecules of the KDO, attached to the disaccharide core. The outer core contains more common hexoses (glucose, galactose and N-acetylglucosamine). O-Antigen is a repeating oligosaccharide unit comprised of 2-6 sugar molecules. [4] (Galdiero et al., 2012)
Lipopolysaccharides play a major role in the outer membrane of Gram-negative bacteria cells. LPS contributes greatly to the structural integrity by increasing the overall negative charge and protects the bacterium cell against the action of lipophilic antibodies and bile salts. For example, in E. coli cells, the ompF and ompC porins exclude passage of all hydrophobic molecules and any hydrophilic molecules greater than 700 Daltons of molecular weight. This prevents penetration of the bacteria by bile salts and other toxic molecules from the GI track. It also acts as a barrier to lysozyme and many antimicrobial agents.
Recent development in the molecular biology field suggested that LPS play as a key factor in septic shock. Septic shock is a serious medical condition that occurs when organ damages or injures in response to infections caused by Gram-negative bacteria which leads to low blood pressure and abnormalities in cellular metabolism. The pathophysiological effects caused by Gram-negative bacteria have been ascribed to lipopolysaccharide (LPS), which has also been termed endotoxin because of its toxic effects (Ruggiero et al., 1993) [5]. LPS causes septic shock by interacting with various components of the immune system and primarily with macrophages. The interaction of LPS with components of host’s immune system leads to production of several endogenous mediators that act as the effectors of the septic shock. LPS triggers the abundant secretion of many macrophage cytokines including Tumour necrosis factor (TNF-α), interleukin-1 (IL-1) and interleukin-6 (IL-6) which together leads to the pathophysiology of septic shock (Meng and Lowell, 1997) [6].
CD14 is discovered to be the major macrophage cell surface receptor for LPS. Presence of the serum protein LPS-binding protein substantially enhances the high-affinity binding of LPS to CD14. Animal models illustrated an important role played by the CD14 in initiating LPS responses. For example, CD14-deficient transgenic mice showed no responses to low dose LPS stimulation while mice overexpressing CD14 were hypersensitive to LPS. Therefore, this indicates that, at higher LPS concentrations macrophages may be activated by a CD14-independent pathway since the CD14 mAbs do not block all biological responses (Meng and Lowell, 1997) [7]. LPS activation of the CD14-TLR4-MD2 complex results in TNF- α expression and tissue factor. The signalling pathways that positively regulate TNF- and TF gene expression in LPS-stimulated macrophage are well characterised. However, mechanisms and signalling pathways that limit the magnitude of the induction of these genes are poorly understood (Guha and Mackman, 2002) [8]. LPS stimulation of macrophages activates the PI3K pathway but the step between the CD14-TLR4-MD2 complex and activation of PI3K has not been solved. HcK, Fgr and Lyn are members of Src-family of PTKs are discovered to be solid candidates for the primary signal transducers of LPS response in macrophages. As a result of LPS treatment, HcK, Fgr and Lyn are activated rapidly. A portion of Lyn directly co-associates with CD14 and PI3-kinase. Expression in macrophages of a constitutively active mutant of Hck augments production of TNF- α while antisense oligonucleotides to Hck inhibit LPS-induced responses. Therefore, protein tyrosine kinases Hck, Fgr, and Lyn play critical roles in LPS-initiated signalling pathways.
Proteomics is the large scale study of proteomes. A proteome is a set of proteins produced in an organism, system or biological context. The main purpose of proteomics is that many types of information cannot be obtained from gene study alone. For example, proteins are responsible for the phenotypes of cells. It becomes impossible to decode the mechanism of ageing, diseases solely by genome study. Therefore, only through the protein studies, it will be possible to decode the mechanism of aging and diseases. Proteomics is used to investigate the rates of protein production, degradation and steady-state abundance, how proteins are modified, how proteins interact with other proteins and the movement of proteins in metabolic pathways and between subcellular compartments.
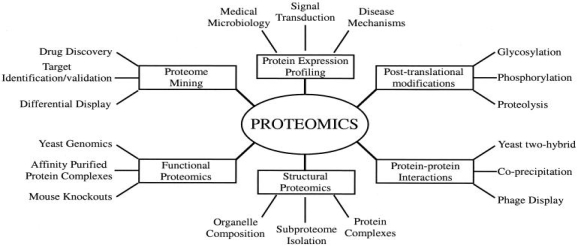
Figure 3: Types of proteomics and their applications to biology. Generally, there are six types of proteomics. They include, structure, protein-protein interactions, proteome mining, post translational modifications, protein expression profiling and functions of proteins of interest. All types of proteomics can be collected using the proteomics experiments (Graves and Haystead, 2002) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC120780/ [9]
To investigate proteomics in more details, several high-throughput technologies have been introduced over the decade. The most commonly used are mass spectroscopy (MS) – based techniques such as Tandem-MS and differential in-gel electrophoresis (DIGE). These methods produce huge amount of data to analyse proteomes (EMBL-EBI Train online, 2018) [10]. Especially, in structural proteomics, X-ray crystallography and nuclear magnetic resonance spectroscopy (NMRS) are used. Structural analysis can help to identify newly discovered genes, show where exactly the drugs bind to proteins and where proteins interact with each other. Protein-protein interactions are used to discover the protein functions and show how proteins are fitted in much larger complexes. Databases are very important for recording and storing the generated data. European Bioinformatics Institute (EBI) allows access to accurate and up-to-date database which allows understanding the link between the results and existing knowledge. There are four significant databases linked to proteomics research. They include, UniprotKB, PRIDE, Reactome and IntAct. UniprotKB is a databse of protein sequences and functional information and databases can be drawn from gene sequence data such as Ensembl and annotation tools such as InterPro (EMBL-EBI Train online, 2018) [11].
Stable isotope labelling with amino acids in cell culture (SILAC) is a general method for mass spectrometric (MS) quantitative proteomics based on metabolic incorporation of stable isotope-labelled amino acids into the cellular protein pool (Lanucara and Eyers, 2011) [12].
https://en.wikipedia.org/wiki/Stable_isotope_labeling_by_amino_acids_in_cell_culture
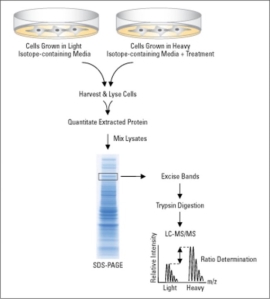
Figure 4: SILAC workflow. SILAC relies on metabolic incorporation of a given light or heavy form of the amino acid into two samples. SILAC requires growing mammalian cells in specialised media deficient in arginine and lysine. This deficiency is compensated by adding “heavy” and “light” forms of the missing amino acids such as 12C6 and 13C6 L-lysine. A SILAC experiment involves growing one cell population in medium containing “heavy” amino acids (experimental), while the other population is grown in the presence of “light” amino acids (control). Both the light and heavy amino acids are incorporated into proteins through natural cellular protein synthesis. Equal numbers of cells from both “heavy” and “light” samples are combined after alteration of the proteome in one sample through genetic manipulation or chemical treatment. Samples are separated by SDS-PAGE and digested with trypsin and finally the samples are analysed using LC-MS/MS. (Thermofisher.com, 2018) [14].
Therefore, the main aim of the report is to use the provided data from LC-MS/MS analysis and identify the signalling pathways activated in response to treating mouse macrophage cell line 264.7 with Lipopolysaccharides (LPS).
Methods and Materials
Mouse macrophage cell line raw 264.7 is established from mouse tissue that originates from Abelson murine leukaemia virus-induced tumour. The purpose of choosing a particular type of tissue is for disease or protein assessment, or retroviral vector production and cell culture support. Treating the raw 264.7 cell line with LPS for 2 days would stimulate lysis of erythrocytes but not tumour cell targets (Lgcstandards-atcc.org, n.d.) [15].
Cell culturing
Mouse macrophage cell line RAW264.7 was grown as two identical cultures on D-MEM + 10% fetal calf serum(FCS) + 1% PenStrep oder RPMI 1640 + 2Mm L-glutamine at 37°C and 5% CO2 and split every second day. The cells were grown to about 80% confluency, detached, washed with phosphate-buffered saline (PBS) and stored at -80°C as described in (Metodieva et al, 2013) [16]
SILAC labelling
Two cell populations are grown in culture media that are identical except that one of the cultures was labelled ‘heavy’ with heavy arginine and lysine and the other culture was labelled ‘light’ with light arginine and lysine. The ‘heavy’ culture was treated with Lipopolysaccharide for 30 minutes. Lipopolysaccharides were not added to the ‘light’ culture as this was our control.1.2×105 cells adapted to grow in D-MEM media into two tissue culture plates with one containing ‘light’ SILAC media and one containing ‘heavy’ media. Both cell populations passaged for at least five cell doublings by splitting cells as appropriate every 2-3 days. Density of cells was maintained so that cells are actively growing in log phase (80% confluency). After five cells doubling, incorporation of heavy arginine and lysine was less than 95%. 106 cells were taken from both ‘light’ and ‘heavy’ samples to determine the incorporation efficiency. After determining the full isotope incorporation, the ‘light’ and ‘heavy’ labelled cells were expanded to find the cell number required for subsequent cell treatment and lysis. Then, the ‘heavy’ culture was treated with 100 units per ml of Lipopolysaccharides (LPS) for 30 minutes. Equal amount of proteins from both cell populations were harvested and combined (Thermofisher.com, 2018) [14].
Protein digestion and preparations of samples for mass spectrometry
The protein samples were mixed with 2×sodium dodecyl sulfate (SDS) sample buffer which was alkylated and reduced and subject to in-gel tryptic digestion kit. The SILAC –labelled proteins were separated into up to 10 fractions by a semi-preparative PAGE before digestion as described in Metodieva et al, 2013.
Isolation of phosphopeptides
Aliquots containing about 0.5 mg of total proteins were separated by a semi-preparative page on single-well minigel. The tryptic peptides were extracted from the gel pieces and 10% of the extracted proteins were used for the quantitative analysis of protein abundance and dried in a vacuum concentrator. The remaining 90% peptides were dissolved in 100μl of 80% acetonitrile containing 2% formic acid and used to isolate phosphorylated peptides. Using the Magnetic TiO2 Phosphopeptide Isolation Kit, the phosphopeptides were isolated. (Metodieva et al, 2013) [16]
LC-MS/MS Analysis and Quantitation of SILAC Peptides
LC-MS/MS analysis was performed using the Hybrid LTQ/Orbitrap Velos instrument from Thermo Fisher (interfaced to a split-less nanoscale). The LTQ/Orbitrap Velos was operated in the Top 20 data-dependent mode where it first executes two high-resolution scans at a resolution of 30,000 (at 400 m/z) followed by 20 MS/MS scans for the 20 most abundant peptide ions having a charge state > 1 as described in Metodieva et al, 2013 [16].
Data Analysis
MS/MS data were analysed by MaxQuant as described in Metodieva et al and further evaluated by statistical tests using Microsoft Excel. The following steps were used to determine the upregulated peptides.
Log transformation of the Heavy/Light ratios
To make the data distribution more normal, H/L ratios were log transformed. Log2 is used for interpretation of the result generated. In Excel spreadsheet, formula =LOG(X, 2) was used where X is the H/L ratio to be transformed. After determining the LOG2 values using =LOG(X, 2), the log values were sorted in the “ascending” order.
Calculation of robust Z scores
Median, P1 and P3 values must be calculated to determine the robust Z scores. The formulas shown below are used to determine Median, P1 and P3 values.
M(median) = MEDIAN (G2:G810) G- log2 value
P1= PERCENTILE (G2:G810, 0.1587) P1 range- 15.87
P3= PERCENTILE (G2:G810, 0.8314) P3 range- 84.13
Using the formula given above, the median was determined to be 0.206268.
P1 was determined to be -0.33414.
P3 was determined to be 0.774425.
Using the determined Median, P1 and P3 values, the robust Z scores were calculated. For the negative log2 values, Z= (X-M)/ (M-P1) was used where (X= (-) log2 value). For the positive log2 values, Z= (X-M)/ (P3-M) was used where (X= (+) log2 value)
Raw P-values and significant peptides calculation using Benjamini and Hochberg FDR method.
P value was determined using the P= 0.5*ERFC (ABS (Z)/SQRT (2)) formula where Z is the calculated robust Z value. The calculated P values were sorted in ascending order for ranking. The peptides were ranked using the Rank= RANK (Z, $I$2: $I$810, 2) where Z is the Z score and $I$2: $I$810 is the range of Z scores in absolute coordinates. Using the rank values, Q values were calculated. The formula was Q= (rank/n) *alpha where rank is the rank value, n is the number of peptides in the data (809) and alpha is the threshold of significance, which was chosen to be 0.05.
Significant peptides selection
The significant peptides were selected using the sig =IF (p
formula where p is the ep value, Q is the Q value. The rows which have 1 in the sig column and positive Z values were the upregulated phosphopeptides (shown in Table 1). The first identifiers were extracted for each upregulated peptide row from the uniprot column. The first identifiers of the peptides are also shown in table 2.
,>
Table 1: 41 peptides were identified as upregulated. Table contains upregulated genes with protein names, uniprot, peptide sequence, modifications, ratio of H/L, log2 value and P values.
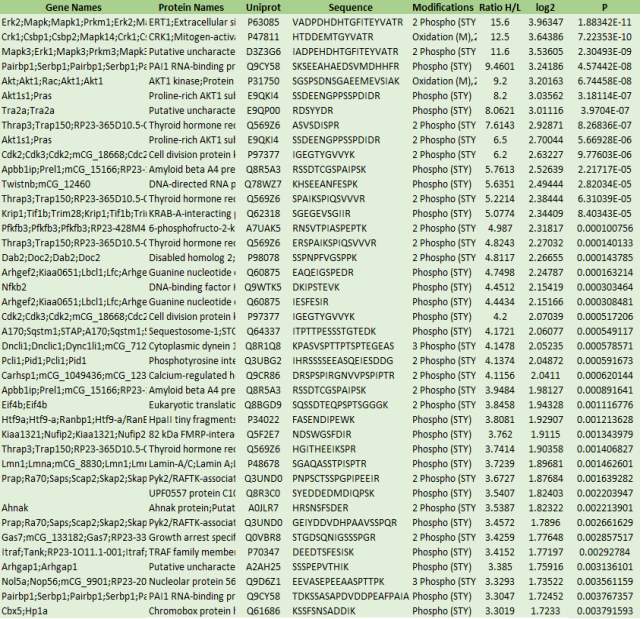
Table 2 above shows that 41 proteins were determined to be upregulated proteins. The peptides with 1 in significant column and have a positive Z values were determined as the upregulated peptides. The uniport ID of 41 peptides were used for the KEGG pathway analysis and the enriched KEGG pathways are shown in figure 5, 6, 7 and 8 respectively.
Pathway analysis
Using the uniport identifiers, the pathways were analysed. The software used for the pathway analysis was webgastalt 2013. Webgastalk 2013 is available at (http://www.webgestalt.org/webgestalt_2013/). The selected organism of interest was “mmusculus” and the gene ID type was “mmusculus_uniprot_swissprot_accession”. Then, the extracted first identifiers were uploaded to the gene list section. The KEGG analysis was selected as the enrichment analysis. Finally, the selected reference set for enrichment analysis was mmusculus_genome and the KEGG enrichment analysis was allowed to run.
RESULTS
Table 3: The table lists the enriched KEGG pathways, number of Entrez IDs in the data set for the pathway, the corresponding Entrez IDS and the statistics for the enriched pathway. Data obtained from http://www.webgestalt.org/webgestalt_2013/htdocs/final_KEGG_file_1515504079.html
The statistic column lists:
C: the number of reference genes in the category
O: the number of genes in the gene set and in the category
E: the expected number in the category
R: ratio of enrichment
rawP: p value from hypergeometric test
adjP: p value adjusted by the multiple test adjustment
| Pathway Name | Gene | EntrezGene | Statistics | |
| Osteoclast differentiation | 5 | 26413 26416 11651 1841218034 | C=118; O=5; E=0.06; R=84.54; rawP=3.70e-09; adjP=1.29e-07 | |
| Progesterone-mediated oocyte maturation | 4 | 26413 26416 12566 11651 | C=88; O=4; E=0.04; R=90.69; rawP=1.15e-07; adjP=2.01e-06 | |
| MTOR signalling pathway | 3 | 26413 11651 75705 | C=53; O=3; E=0.03; R=112.94; rawP=2.61e-06; adjP=3.05e-05 | |
| MAPK signalling pathway | 4 | 26413 26416 11651 18034 | C=268; O=4; E=0.13; R=29.78; rawP=9.76e-06; adjP=5.69e-05 | |
| VEGF signalling pathway | 3 | 26413 26416 11651 | C=76; O=3; E=0.04; R=78.76; rawP=7.76e-06; adjP=5.69e-05 | |
| Fc epsilon RI signalling pathway | 3 | 26413 26416 11651 | C=80; O=3; E=0.04; R=74.82; rawP=9.06e-06; adjP=5.69e-05 | |
| Prostate cancer | 3 | 26413 12566 11651 | C=89; O=3; E=0.04; R=67.25; rawP=1.25e-05; adjP=6.25e-05 | |
| Toll-like receptor signalling pathway | 3 | 26413 26416 11651 | C=101; O=3; E=0.05; R=59.26; rawP=1.82e-05; adjP=7.08e-05 | |
| Chagas disease (American trypanosomiasis) | 3 | 26413 26416 11651 | C=100; O=3; E=0.05; R=59.86; rawP=1.77e-05; adjP=7.08e-05 | |
| Pathways in cancer | 4 | 26413 12566 11651 18034 | C=325; O=4; E=0.16; R=24.56; rawP=2.08e-05; adjP=7.28e-05 | |
The table 3 above shows that mapk1/mapk3 (26413) and akt1 (11651) are present in all the enriched KEGG pathways. mapk14 (26416) is present in 7 out of 10 enriched pathways. Nfkb2 (19034) and cdk2 (12566) are mapped onto 3 out of 10 signalling pathways. Sqstm1 (18421) is present only in the osteoclast differentiation pathway and eif4b (75705) is only present in the mTOR signalling pathway. Therefore, this data indicates mapk activation, akt activation and possibly some element of toll-like receptor signalling pathway as well because map kinases and akt feature in all of the enriched pathways.
325 genes from the data were mapped onto the cancer signalling pathway. There were 118 reference genes present in the osteoclast differentiation and 5 genes were mapped onto the signalling pathway. The expected number in the category was 0.06 which is higher than progesterone-mediated oocyte maturation, mTOR, VEGF, FC epsilon RI, prostate cancer and Chagas disease. However, there were 286 genes present in the MAPK signalling pathway and the E value was 0.13. Therefore, higher the E value, higher the chance of genes mapping onto the signalling pathways if it was random. MTOR signalling pathway has the highest ratio of enrichment compared to other enriched pathways and at the same time it has the lowest E value of 0.03. Therefore, the chances of proteins mapping to the mTOR signalling pathway is lower than other pathways.
Table 2: 8 peptides were identified as significant peptides from the table 1. It contains gene name, uniport accession, peptide sequence, H/L ratio, p-value and phosphorylation sites. Genes highlighted in red codes for the protein that are mapped onto the enriched KEGG pathways. S-497 means phosphorylation at the Serine residue, T-183 means phosphorylation at the threonine residue Y-185 means phosphorylation at tyrosine residue.

Mapk1 features the largest H/L ratio of 15.6 whereas EiF4b features the lowest H/L value of 3.8458. The highest p- value of 0.001116 belongs to Eif4b. Mapk1, mapk3 and mapk14 were discovered to be dually phosphorylated. Mapk1 gets phosphorylated on the residues threonine-183 and tyrosine-185 by MAP2K1 and MAPK2K. Mapk3 is phosphorylated on the residues threonine-203 and tyrosine-205 by MAP2K1 and MAP2K2. However, Mapk14 is phosphorylated on the residues threonine-180 and tyrosine-182 by three kinases (MAP2K3, MAP2K4 and MAP2K6). Cdk2 is phosphorylated by WEE1 at the tyrosine-15 residue and threonine-14 and tyrosine-19 are phosphorylated by unknown proteins. No phosphorylation takes place on the given nfkb2 peptide. Eif4b features five phosphorylation sites and they are phosphorylated by unknown proteins.
The enriched KEGG Osteoclast differentiation pathway
Table 4: Genes in the Osteoclast differentiation pathway are listed. For each gene, the table lists the Entrez ID, Gene Symbol, the description and proteins coded by the genes. Entrez Gene ID is linked to the Entrez Gene databases, respectively. Data is obtained from KEGG database.
(http://www.kegg.jp/pathway/mmu04380+26413+26416+11651+18412+18034)
| Gene symbol | Gene name | EntrezGene | Proteins coded by the gene |
| Mapk1 | Mitogen-activated protein kinase 1 | 26413 | ERK |
| Mapk14 | Mitogen-activated protein kinase 14 | 26416 | P38 |
| Akt1 | Thymoma viral proto-oncogene 1 | 11651 | Akt |
| Sqstm1 | Sequestosome 1 | 18412 | P62 |
| Nfbk2 | Nuclear factor of kappa light polypeptide gene enhancer in B cells 2, p49/p100 | 18034 | NFkB |
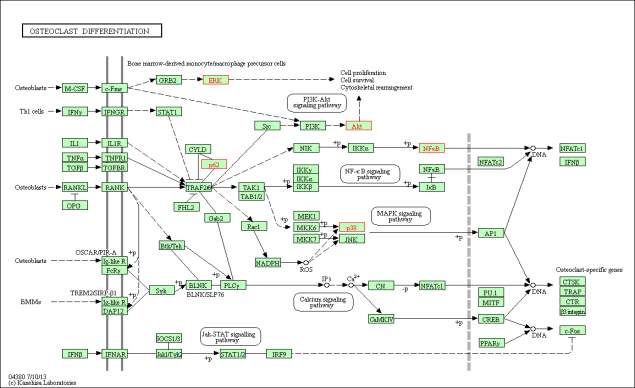
Figure 5: The enriched KEGG Osteoclast differentiation pathway. The proteins that are highlighted in red are the proteins mapped onto the pathway from the upregulated peptide list. The mapped proteins are ERK, p38, Akt, p62 and NFkB. The genes coding for the mapped proteins are shown in table 4. Phosphorylation of ERK leads to activation of cell proliferation, cell survival and cytoskeletal management. Phosphorylation of AKT by PIK3 also leads to activation cell proliferation (PI3K-Akt signalling pathway). NfkB and p38 phosphorylation leads to activation of NFATc1.
The enriched KEGG MAPK signalling pathway
Table 5: Genes in the MAPK signalling pathway are listed. For each gene, the table lists the Entrez ID, Gene Symbol, the description and proteins coded by the genes. Entrez Gene ID is linked to the Entrez Gene databases, respectively. Data is obtained from KEGG database. (http://www.webgestalt.org/webgestalt_2013/htdocs/final_sig_kegg_file_1515504079.html#MAPK signaling pathway)
| Gene symbol | Gene name | EntrezGene | Proteins coded by the gene |
| Mapk1 | Mitogen-activated protein kinase 1 | 26413 | ERK |
| Mapk14 | Mitogen-activated protein kinase 14 | 26416 | p38 |
| Akt1 | Thymoma viral proto-oncogene 1 | 11651 | AKT |
| Nfkb2 | nuclear factor of kappa light polypeptide gene enhancer in B cells 2, p49/p100 | 18034 | NfkB |
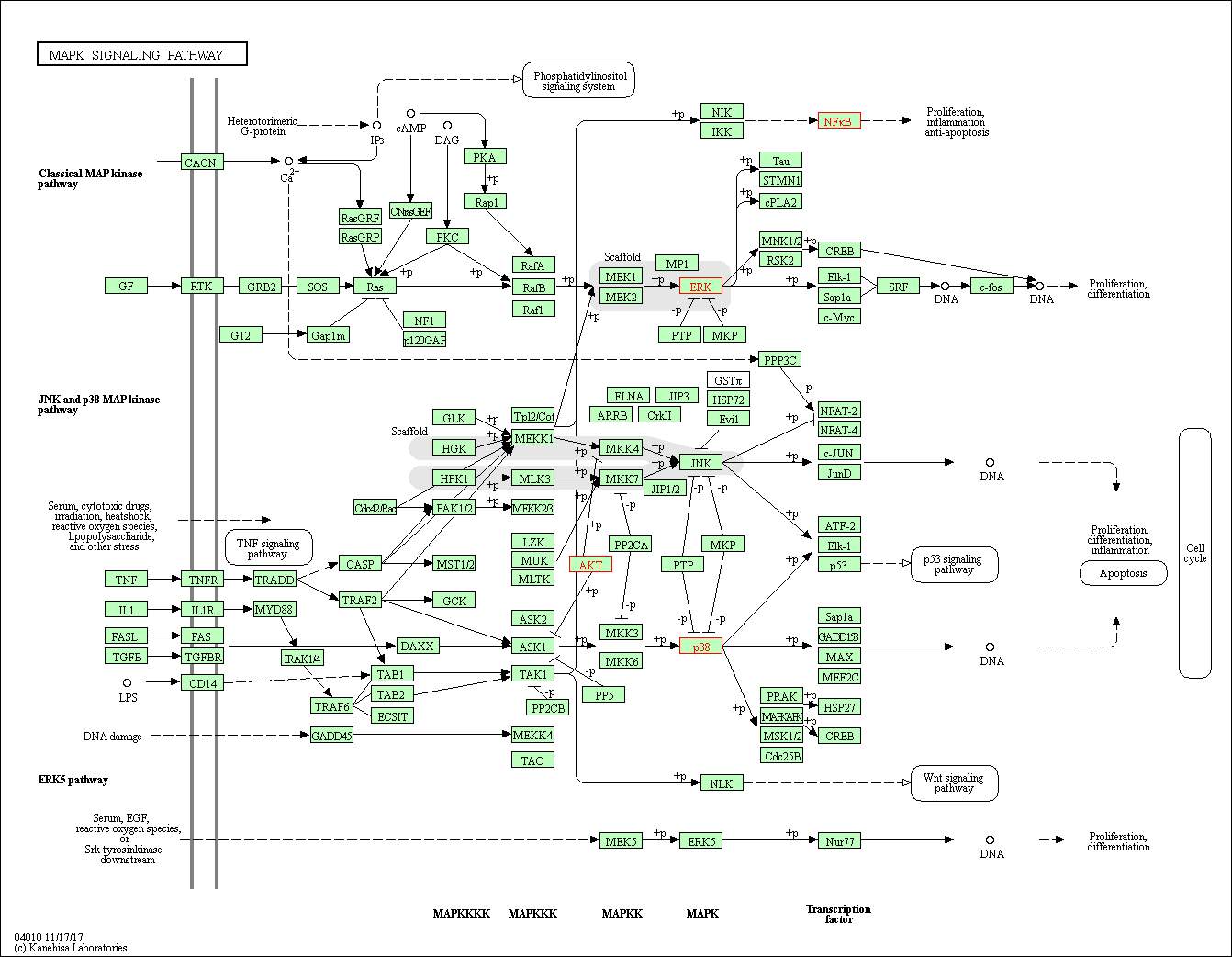
Figure 6: The enriched KEGG MAPK signalling pathway. The proteins highlighted in red are the proteins mapped onto the pathway from the upregulated peptide list (table 1). The mapped proteins are ERK, p38, Akt. The genes coding for the mapped proteins are shown in table 4. Phosphorylation of p38 by Map2k3 and map2k6 leads to phosphorylation of ATF-2, Elk01 and p53 proteins and activates the p53 signalling pathway. Phosphorylation of ERK by map2k1 and map2k2 leads to phosphorylation of Tau, STMN1, CPLA2, MNK1/2 and RSK2 by ERK. RSK2 and MNK1/2 phosphorylates CREB. Therefore, phosphorylation of CERB leads to activation of proliferation differentiation
The enriched KEGG Toll-like receptor signalling pathway
Table 6: Genes in the Toll-like receptor signalling pathway are listed. For each gene, the table lists the Entrez ID, Gene Symbol, the description and proteins coded by the genes. Entrez Gene ID is linked to the Entrez Gene databases, respectively. Data is obtained from KEGG database. (http://www.webgestalt.org/webgestalt_2013/htdocs/final_sig_kegg_file_1515504079.html#Toll-like receptor signaling pathway)
| Gene symbol | Gene name | EntrezGene | Proteins coded by the gene |
| Mapk1 | Mitogen-activated protein kinase 1 | 26413 | ERK |
| Mapk14 | Mitogen-activated protein kinase 14 | 26416 | p38 |
| Akt1 | Thymoma viral proto-oncogene 1 | 11651 | AKT |
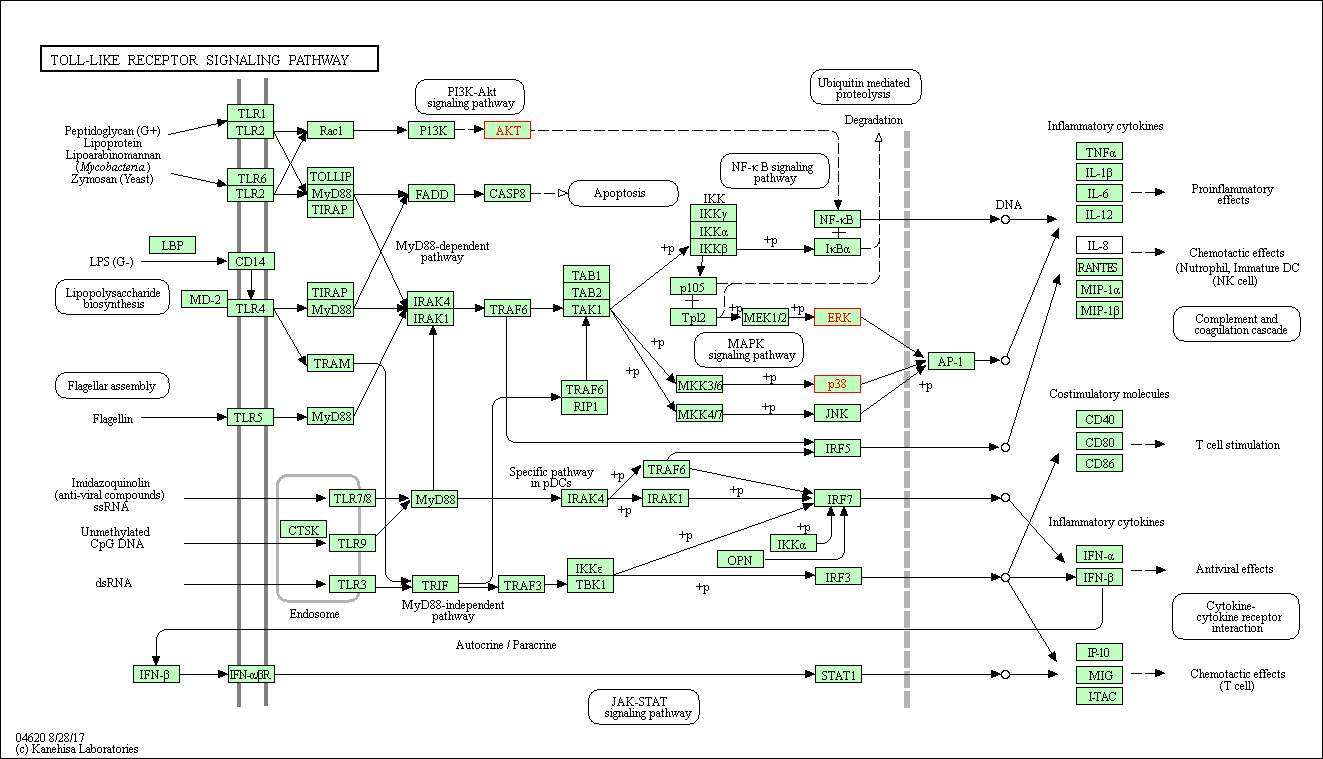
Figure 7: the enriched KEGG Toll-like receptor signalling pathway. The proteins highlighted in red are mapped onto the signalling pathway. The mapped on proteins are AKT, ERK and p38. The genes coding for the proteins are shown in table 6. LPS binds to the transmembrane protein CD14 or TLR4 and phosphorylates TIRAP and MyD88. TIRAP and MyD88 lead to activation of FADD and FADD activates CSP8. This activation results in cell apoptosis. TIRAP and MYD88 activate IRAK4 and IRAK1 and they phosphorylate IKK subunits which activates the P105 and TP12 proteins. P105 and TP12 activation leads to phosphorylation of ERK protein. AKT is activated by the PI3K and it leads to activation of NF-kb which leads to activation of inflammatory cytokines such as TNF-α, IL-1β, IL-6 and IL-12. Activation of cytokines leads to pro inflammatory effects. Both ERK and p38 phosphorylates AP-1 and leads to activation of inflammatory cytokines. LPS bni
Discussion
The uniport IDs of the 41 significant peptides were used to identify the enriched KEGG pathways using webgestalt 2013. 10 different signalling pathways were obtained from the KEGG pathway analysis. The KEGG enriched pathways were osteoclast differentiation, progesterone-mediated oocyte maturation, mTOR, MAPK, VEGF, Fc epsilon RI, prostate cancer, Toll-like receptor, chages disease and pathways in cancer. However, only Osteoclast differentiation, MAPK and Toll-like receptor signalling pathways were selected for further analysis. This is because, there are 5 genes from the significant peptides list (table 1) were mapped onto the osteoclast differentiation. Also, 118 reference genes in the category was mapped onto the pathway and the expected number in the category (E) was 0.06 which was higher than progesterone-mediated (E= 0.04), VEGF (E= 0.04), Fc epsilon RI (E= 0.04), prostate cancer (E= 0.04) and Chagas disease (E= 0.05). MAPK signalling pathway was also chosen because it has 268 genes from the category and the E value was 0.13. Toll-like receptor signalling pathway was also chosen because many studies show that TLR4 plays a crucial role in LPS recognition. Stimulation of Toll-like receptor 4 by LPS induced the critical pro inflammatory cytokines that are necessary to achieve the potent immune response (Lu, Yeh and Ohashi, 2008) [17].
The information obtained from the enriched KEGG pathway analysis is highly rebundant. This means, identical subset of proteins were mapped to multiple enriched pathways. For example, VEGF, Fc epsilon RI, prostate cancer, pathways in cancer and Chagas disease signalling pathways have mapk1, mapk14 and akt1 proteins mapped on to the pathway. Therefore, these pathways were ignored and MAPK, Toll-like and osteoclast differentiation pathways were selected as the pathways activated in response to treating mouse cells with LPS.
To prove that osteoclast differentiation, MAPK and Toll-like receptor signalling pathways were activated in response to treating cells with LPS, the phosphorylation sites of the proteins mapped onto the pathway were analysed. In the osteoclast differentiation pathway, the proteins mapped onto the pathway were ERK (mapk1/mapk3), p38 (mapk14), P62 (sqstm1), AKT (akt1) and NFKB (Nfkb2). ERK, p38, AKT and NFKB were mapped on to the MAPK signalling pathway. ERK, p38 and AKT proteins were mapped on the Toll-like receptor pathway.
Mapk1 is one of the proteins activated because Mapk1 is activated by dual phosphorylation which changes the conformation shape that activates the kinase. In the data file, the given peptide sequence of the mapk1 was VADPDHDHTGFITEYVATR and it is found to be phosphorylated on two residues (STY). This shows that activation of Mapk1 occurs through phosphorylation of threonine at 183 and tyrosine residues at 185 at the sequence TEY by upstream MAP kinase kinases, MAP2K1 and MAP2K2 (Uniprot.org, 2018) [18]. Phosphorylation of both the threonine and tyrosine are required for activity. This phosphorylation causes dramatic conformational changes, which enable full activation and interaction of MAPK1/ERK2 with its substrates (Phosphosite.org, 2018) [19].
Similarly, mapk3 is also activated because it is a close homologue to the mapk1 and it gets phosphorylated at the TEY residue like mapk1. The given peptide sequence was IADPEHDHTGFITEYVATR and it is found to be phosphorylated on two residues (STY). This proves that mapk1 can be activated when dual phosphorylation takes place. The dual phosphorylation takes at threonine at 203 and tyrosine at 205 of the given peptide by MAP2K1 and MAP2K2. Therefore, dual phosphorylation on the given peptide of mapk1 and mapk3 proves that our experimental results are strongly upregulated which is reflected by the heavy to light ratio 15.6 and 11.6 times heavy forms more abundant than the light form. Therefore, this proves that mapk1/ mapk3 pathways were activated.
Mapk14 is another protein mapped onto the osteoclast differentiation pathway. The given peptide sequence of mapk14 was HTDDEMTGYVATR and it found to be phosphorylated on two residues and oxidised on the M residue. Dual phosphorylation takes place at the Threonine-180 and tyrosine-182 by MAPK2K3, MAP2K4 and MAP2K6. Dually phosphorylated on thr-180 and try-182 by the MAPK2K3, MAP2K4 and MAP2K6 in response to inflammatory cytokines, growth factors and environmental stress which activates the enzyme (Uniprot.org, 2018) [20]. Therefore, dual phosphorylation on the given peptide proves that our experimental results are strongly upregulated which is reflected by the heavy to light ratio of 12.5 shows that the heavy forms are 12.5 times more abundant than the light form. Therefore, this proves mapk14 pathways were activated.
The given Akt1 sequence is SGSPSDNSGAEEMEVSLAK and it is found to be phosphorylated on two residues for activation. Akt1 is activated by dual phosphorylation. Phosphorylation takes place on the Threonine-124 and threonine-126. Therefore, dual phosphorylation on the given peptide shows that our experimental results were upregulated, reflected by the heavy to light ratio of 9.2 shows that heavy forms are 9.2 times more abundant, thus proving that akt1 was activated in the signalling pathway.Akt1 an oncogenic AGC kinase that plays an important role in regulating cell survival and metabolism in many different signalling pathways. Phosphorylation alos takes place on the T-308.T308 is phosphorylated by PDK1 in the PI3 kinase pathway, and S473 is phosphorylated by mTOR in the mTORC2 pathway. The ‘Lys-63’-linked ubiquitination of AKT1 by TRAF6 is important for its translocation to the plasma membrane, phosphorylation, and activation. When Akt is fully phosphorylated, it translocate into the nucleus, undergoes ‘Lys-48’-polyubiquitination catalysed by TTC3, leading to its proteosomal degradation. Hyperactive or overexpressed in many cancers including breast, prostate, lung, pancreatic, liver, ovarian and colorectal. (uniport.org, 2018)[20]
The transcription factor NFkB gets phosphorylated by MAP3K14 at S-866 and S-870. This shows NFkB does not get phosphorylated on the given peptide sequence. This is proven by the heavy to light ratio of 4.4512. Therefore, lower heavy to light ratio means there is a lower chance of phosphorylation taking place on the given peptide. The data suggests that p62 protein is phosphorylated once on the given peptide. However, analysed proved that there were two phosphorylation on the given peptide, threonine-269 and threonine-272 and the phosphorylating proteins are unknown. The p62 protein in osteoclast differentiation phosphorylates CYLD protein at Threonine-272. Therefore, it is proven that the pathways activated in response to treating cells with LPS are osteoclast differentiation, MAPK and Toll-like receptor signalling pathways.
References
Han, W., Wu, B., Li, L., Zhao, G., Woodward, R., Pettit, N., Cai, L., Thon, V. and Wang, P. (2011). Defining Function of Lipopolysaccharide O-antigen Ligase WaaL Using Chemoenzymatically Synthesized Substrates. Journal of Biological Chemistry, [online] 287(8), pp.5357-5365. Available at: http://www.jbc.org/content/287/8/5357.full [Accessed 1 Jan. 2018].
Dreamstime.com. (2018). Gram-positive And Gram-negative Bacteria Stock Vector – Image: 45337024. [online] Available at: https://www.dreamstime.com/stock-illustration-gram-positive-gram-negative-bacteria-difference-bacterial-image45337024 [Accessed 3 Jan. 2018].
Galanos, C., Lüderitz, O. and Westphal, O. (2018). Lipopolysaccharides – Structure, Function and Application. [online] Sigma-Aldrich. Available at: https://www.sigmaaldrich.com/technical-documents/articles/biology/glycobiology/lipopolysaccharides.html [Accessed 4 Jan. 2018].
Galdiero, S., Falanga, A., Cantisani, M., Tarallo, R., Elena Della Pepa, M., D’Oriano, V. and Galdiero, M. (2012). Microbe-Host Interactions: Structure and Role of Gram-Negative Bacterial Porins. Current Protein and Peptide Science, [online] 13(8), pp.843-854. Available at: https://www.researchgate.net/publication/234103523_Microbe-Host_Interactions_Structure_and_Role_of_Gram-Negative_Bacterial_Porins [Accessed 4 Jan. 2018].
Ruggiero, V., D’Urso, C., Albertoni, C., Campo, S., Foresta, P. and Martelli, E. (1993). LPS-induced serum TNF production and lethality in mice: effect of L-carnitine and some acyl-derivatives. Mediators of Inflammation, [online] 2(7), pp.S43-S50. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2365448/pdf/MI-02-S453.pdf [Accessed 5 Jan. 2018].
Meng, F. and Lowell, C. (1997). Lipopolysaccharide (LPS)-induced Macrophage Activation and Signal Transduction in the Absence of Src-Family Kinases Hck, Fgr, and Lyn. The Journal of Experimental Medicine, [online] 185(9), pp.1661-1670. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2196288/ [Accessed 5 Jan. 2018].
Meng, F. and Lowell, C. (1997). Lipopolysaccharide (LPS)-induced Macrophage Activation and Signal Transduction in the Absence of Src-Family Kinases Hck, Fgr, and Lyn. The Journal of Experimental Medicine, [online] 185(9), pp.1661-1670. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2196288/ [Accessed 5 Jan. 2018].
Guha, M. and Mackman, N. (2002). The Phosphatidylinositol 3-Kinase-Akt Pathway Limits Lipopolysaccharide Activation of Signaling Pathways and Expression of Inflammatory Mediators in Human Monocytic Cells. Journal of Biological Chemistry, [online] 277(35), pp.32124-32132. Available at: http://www.jbc.org/content/early/2002/06/06/jbc.M203298200.full.pdf [Accessed 5 Jan. 2018].
Graves, P. and Haystead, T. (2002). Molecular Biologist’s Guide to Proteomics. Microbiology and Molecular Biology Reviews, [online] 66(1), pp.39-63. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC120780/ [Accessed 6 Jan. 2018].
EMBL-EBI Train online. (2018). What is proteomics?. [online] Available at: https://www.ebi.ac.uk/training/online/course/proteomics-introduction-ebi-resources/what-proteomics [Accessed 8 Jan. 2018].
Mestrovic, D. (2016). What is Proteomics?. [online] News-Medical.net. Available at: https://www.news-medical.net/life-sciences/What-is-Proteomics.aspx [Accessed 12 Jan. 2018].
Lanucara, F. and Eyers, C. (2011). Mass Spectrometric-Based Quantitative Proteomics Using SILAC. Methods in Enzymology, [online] pp.133-150. Available at: https://www.ncbi.nlm.nih.gov/pubmed/21943896 [Accessed 10 Jan. 2018].
O
Thermofisher.com. (2018). SILAC Protein Quantitation Kit – DMEM:F12 – Thermo Fisher Scientific. [online] Available at: https://www.thermofisher.com/order/catalog/product/88439 [Accessed 12 Jan. 2018].
Lgcstandards-atcc.org. (n.d.). RAW 264.7 ATCC® TIB-71™. [online] Available at: https://www.lgcstandards-atcc.org/Products/All/TIB-71.aspx [Accessed 12 Jan. 2018].
Metodieva, G., Nogueira-de-Souza, N., Greenwood, C., Al-Janabi, K., Leng, L., Bucala, R. and Metodiev, M. (2013). CD74-dependent Deregulation of the Tumor Suppressor Scribble in Human Epithelial and Breast Cancer Cells. Neoplasia, 15(6), pp.660-IN21.
Lu, Y., Yeh, W. and Ohashi, P. (2008). LPS/TLR4 signal transduction pathway. Cytokine, [online] 42(2), pp.145-151. Available at: https://www.ncbi.nlm.nih.gov/pubmed/18304834 [Accessed 9 Jan. 2018].
Uniprot.org. (2018). Mapk1 – Mitogen-activated protein kinase 1 – Mus musculus (Mouse) – Mapk1 gene & protein. [online] Available at: http://www.uniprot.org/uniprot/P63085 [Accessed 15 Jan. 2018].
Phosphosite.org. (2018). ERK2 (mouse). [online] Available at: https://www.phosphosite.org/proteinAction.action?id=1338&showAllSites=true [Accessed 15 Jan. 2018].
Uniprot.org. (2018). MAPK14 – Mitogen-activated protein kinase 14 – Homo sapiens (Human) – MAPK14 gene & protein. [online] Available at: http://www.uniprot.org/uniprot/Q16539 [Accessed 15 Jan. 2018].
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biomedical Science"
Biomedical Science focuses on how cells, organs and systems function in the human body and underpins much of modern medicine. Biomedical Science applies parts of natural and/or formal sciences to help develop advances in healthcare.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: