Detecting Hypertrophic Cardiomyopathy by Screening Programmes
Info: 8991 words (36 pages) Dissertation
Published: 22nd Feb 2022
1 Abstract
Objectives: The aims of this overview was to determine if hypertrophic cardiomyopathy can be detected accurately, reliably and effectively by screening programmes, and if screening programmes are effective in reducing the rate of mortality of sudden cardiac death.
Background: Hypertrophic cardiomyopathy (HCM) is a condition where the myocardium becomes thickened and stiff. It affects approximately 1 in 500 individuals.
The thickening cannot be explained by normal loading conditions. Stiffened cardiac tissue results in a reduced contractile force, and consequently reduced blood exiting the heart. It is usually asymptomatic until it presents disastrously as SCD and an incident is most likely to occur in young athletes. The disease can occur at any age and both genders are affected equally. Common symptoms are shortness of breath, chest pain, palpitations, light headedness and fainting. Palpitations occur due to supraventricular or ventricular arrhythmias. There is no cure and treatments work to manage the symptoms. Surgical procedures like septal myectomy are options to remove the hypertrophied myocardium. The condition is most commonly inherited via familial routes but can also occur due to various other genetic and non-genetic causes. The condition can be easily identified by characteristic traits shown on electrocardiogram (ECG) screening. Echocardiogram screening and cardiac magnetic resonance imaging can be used if ECG screening is inconclusive.
Methods: Pubmed and Cochrane Library were extensively searched to identify systematic reviews which performed investigations into the effectiveness of ECG screening. From 49 reviews, 3 were included for overview.
Results: Data showed that ECG screening had a 94% sensitivity and 93% specificity. Pooled data showed that 94.3% of HCM cases displayed single or multiple ECG abnormalities. The results showed that the prevalence of HCM was lower than reported, at 1 in 835.
Conclusions: Screening programmes have been shown to be able to successfully diagnose HCM and various other conditions associated with SCD. This overview has shown that screening programmes utilising ECG is highly sensitive and specific when diagnosing HCM and other potentially lethal cardiac disorders. If applied on a large scale in the UK, screening programmes have the opportunity to reduce the case-fatality rate of HCM by making asymptomatic individuals aware that they have to condition, and potentially save approximately 240 lives. The NSH should provide screening programmes to identify dangerous, potentially lethal, asymptomatic cardiac conditions such as HCM, to prevent the unnecessary, treatable deaths from SCD of occurring.
Contents
2. Introduction
3. Background
3.1. Hypertrophic cardiomyopathy
3.2. Genetics
3.3. Screening using ECG
3.4. Screening using ECHO
3.5. Screening using cMRI
3.6. Treatment and management
3.7. Surgical interventions
3.8. Other common conditions
4. Methods
5. Results
5.1. Is screening reliable and accurate?
5.2. Does screening reduce the rate of mortality of SCD?
6. Discussion
6.1. Screening as an effective diagnostic tool
6.2. Strengths and limitations
7. Conclusion
8. Acknowledgements
9. References
2 Introduction
Sudden cardiac death (SCD), most usually associated with hypertrophic cardiomyopathy (HCM), affects approximately 1 in 500 individuals in the UK.1 Defined as a “unexpected natural death from a cardiac cause within a short time period, generally ≤1 hour from the onset of symptoms, in a person without any prior condition that would appear fatal”, the condition is usually asymptomatic until a sudden cardiac arrest, an unexpected heart attack in an individual who looks healthy.2 The mortality rate for this condition is somewhere between 1-5%, however the condition is easily diagnosed using electrocardiogram (ECG) and echocardiogram (ECHO) screening.2 This is done by healthcare professionals who firstly conduct an electrocardiogram and identify key abnormalities in the Q, S, T and P waves. If there is reason to believe that HCM is present, but cannot be confirmed from ECG alone, then an ECHO can be used to confirm/deny a diagnosis. ECGs can identify HCM and the other common conditions which cause SCD due to the electrical abnormalities caused by cardiac arrhythmias. The most common arrhythmias are ventricular or atrial, the uncontrolled, disorganised electrical impulse firing from the ventricles or atria, resulting in inability of the heart to pump blood through the body and death can occur within minutes.
Currently in the UK, these screening programmes are not funded by the NHS and individuals must go to external organisations such as Cardiac Risk in the Young (CRY) for screening, unless there is a family history of inherited heart conditions or SCD at a young age. The decision to not fund screening through the NHS was based on a report conducted by the UK National Screening Committee in 2015, which states that the accuracy of the testing service is unassessed, and that “the conditions that lead to sudden cardiac death are poorly understood and there is no evidence to guide clinicians regarding treatment or lifestyle advice when such a problem is found in a family member or when detected at a screening examination”3. These claims are disputed by CRY who claim that the conclusions of the committee are “false and misleading”. The conclusions are also conflicted by chapter eight of the National Service Framework, published in March 2005 which states that “When sudden cardiac death occurs, NHS services have systems in place to identify family members at risk and provide personally tailored, sensitive and expert support, diagnosis, treatment, information and advice to close relatives” and “Sudden cardiac death in younger people is often indicative of inherited cardiac disease. There is real potential to prevent further tragedies by the appropriate care of family members in these cases”4. These reports show clear contradiction with each other, and the reasons for NHS funding to be denied appear to be unsupported and in need of review. This overview of systematic reviews attempts to identify whether the underlying cause of SCD, HCM is screened for correctly and adequately in the UK.
The objectives of this dissertation are:
- To identify how hypertrophic cardiomyopathy can be screened for.
- To identify if screening is an effective method for reducing the rate of mortality from SCD.
- To determine if screening is accurate and reliable in its ability to diagnose HCM.
3 Background
3.1 Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy is a condition where the myocardium becomes thickened and stiff (figure 1).1 This is most commonly found in the left ventricle and septum, leading to obstruction of blood flow into the aorta or pulmonary artery, but it can occur in any region of the heart.1 The thickening cannot be explained by normal loading conditions, meaning the normal wall tensions and stresses occurring in the heart during systole and diastole.5 Stiffened cardiac tissue results in a reduced contractile force, and consequently reduced blood exiting the heart.1 It is usually asymptomatic until it presents disastrously as SCD and an incident is most likely to occur in young athletes due to the increased overall “use” of the heart by this group.1 The disease can occur at any age and both genders are affected equally. If symptoms present, common symptoms are shortness of breath, chest pain, palpitations, light headedness and fainting.6 Exertional dyspnoea is caused by left ventricular outflow tract (LVOT) obstruction associated with increased left ventricle wall hypertrophy or stiffness.7 Exertional syncope/pre-syncope can be caused by dynamic LVOT disruption and/or ventricular dysrhythmia.6 Other common symptoms which are directly associated with hypertrophy are mitral valve regurgitation, ventricular diastolic dysfunction, ventricular systolic dysfunction and chest pain caused by increased blood demand and reduced supply.8,9,10,11 Palpitations occur due to supraventricular or ventricular arrhythmias.12 Although the condition is most commonly inherited via familial routes, 40-60% of cases, it can also occur due to various other genetic and non-genetic causes including inborn errors of metabolism, neuromuscular diseases e.g. Freidreich’s ataxia, mitochondrial diseases e.g. MELAS, malformation syndromes e.g. Noonan’s syndrome, amyloidosis and drug induction e.g. Tacrolimus.5 These other causes only account for 5-10% of cases though, and approximately 25-30% of cases have an unknown cause.5
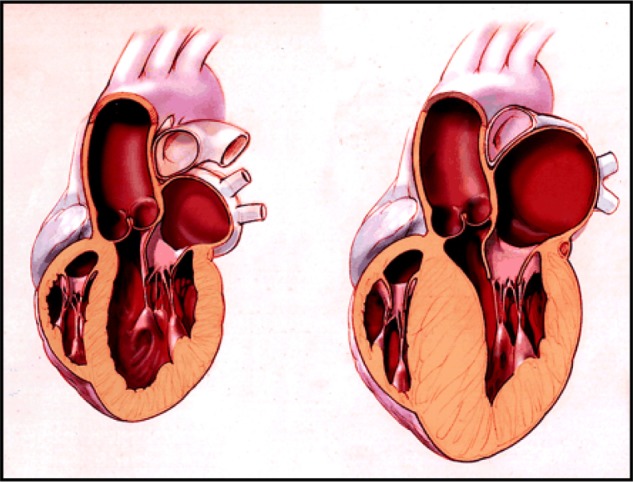
Figure 1. Representations of a normal heart (left) and a heart with HCM (right).1
3.2 Genetics
HCM affects approximately 1 in 500 adults, and it is therefore the most common genetically inheritable cardiovascular disease.13,14 The condition is inherited as an autosomal dominant trait meaning there is a 50% chance of offspring of an affected individual also being affected. It has been associated with 11 or more genes coding for protein constituents of the sarcomere.15 The most common mutations occur in MYH7 and MYBPC3 genes, encoding for β-myosin heavy chains and myosin binding protein C.5 It was originally believed that these mutations would cause sarcomere dysfunction leading to compensatory hypertrophy, but this was argued due to the fact that some develop HCM later in life, and the mutation is present from birth, thus this cannot be explained by a compensatory mechanism.15 The mechanism remains unclear, but it is now believed that mutations in these genes lead to increased calcium sensitivity in the myocytes, causing myocyte dysfunction leading to hypertrophy.15 Other common mutations are found in cardiac troponin genes; TNNI2, TNNI3, TPM1, and myosin light chain 3 (MYL3).16 These genes along with their estimated frequency are shown in Table 1. Patients with any of these mutations usually have higher historical family prevalence of HCM and also symptomatically present earlier than individuals without.17 Phenotype heterogeneity is common, suggesting that single mutations are not the only factor of phenotype or clinical course.18 It appears that a mutation in any of the associated genes have the same clinical outcome removing the need to use genetic testing for prognosis outcomes.19 The genes for α-myosin heavy chain, titin, muscle LIM protein, telethonin, vinculin, and junctophilin 2 have been associated with HCM but more research is required to determine their absolute pathogenicity.16
3.3 Screening using ECG
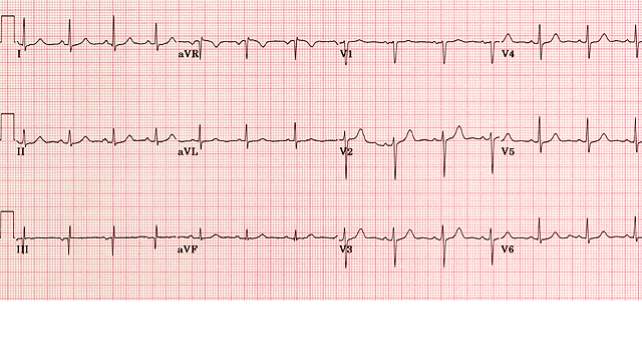
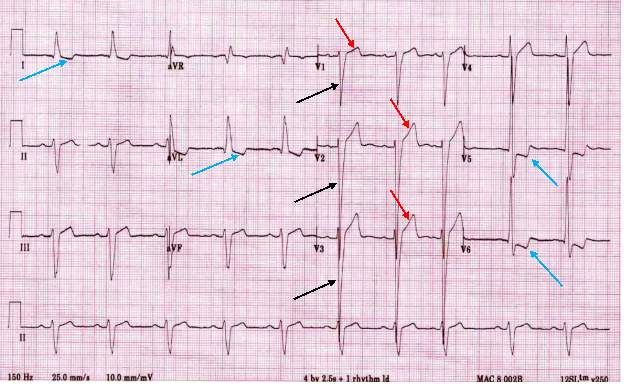
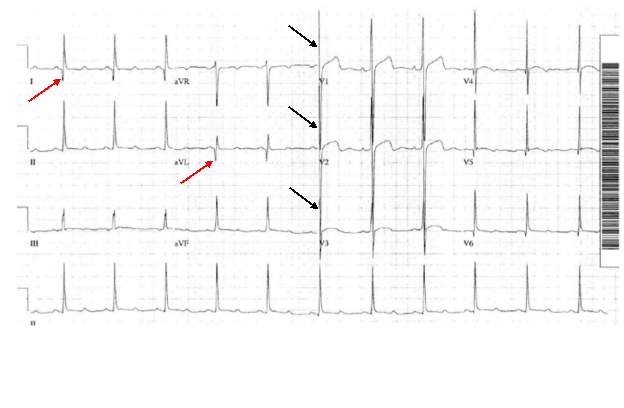
HCM can easily be identified and diagnosed using a 12-lead electrocardiogram.20 12 lead ECG screening involves placing 12 electrodes at various points on the chest (precordial), wrists and ankles. These electrodes record the electrical activity of the heart. The leads V1-6 are the precordial leads. Leads I, II, III, aVL (augmented vector left), aVR (augmented vector right) and aVF (augmented vector foot) are all used to create a vertical plane of the transmission pattern. An ECG of a healthy, normal heart would show the classic and iconic waveform. An ECG scan of a healthy heart is shown in figure 2. However there are many indicators which can indicate a positive HCM diagnosis. Classical ECG trace examples of HCM show high voltages with restricted or extensive repolarization abnormalities.1 If the left ventricle is hypertrophic increased precordial voltages, non-specific ST segment and T-wave abnormalities would be expected, as shown in figure 3. In asymmetrical septal hypertrophy narrow, deep, “dagger-like” Q waves in lateral and inferior leads (I, II and III), where the infarction Q waves are greater than 40ms in duration would be expected, as shown in figure 4. In apical HCM large precordial voltages, giant T wave inversions in the precordial leads and inverted T waves in inferior and lateral leads would be expected, as shown in figure 5. Left ventricle diastolic dysfunction leading to compensatory left atrial hypertrophy would show as P mitrale, shown on the ECG trace as broad, notched P waves, as shown in figure 6.
ECG screening is the first line of screening, due to its simplicity and the ease of the screening procedure. However its cost effectiveness, sensitivity and specificity have been questioned. ECG screening has been introduced in Italy for athletes aged 12 to 35.22 Similarly in the United States, some colleges have introduced pre-participation screening for their athletes, potentially meaning ECG screening implementation for 10 million young athletes, however they are established requirements of the major sports leagues and have been used for over 50 years.23 Now there is debate in the UK as to whether pre-participation screening programmes should be introduced for young athletes, and funded by the NHS rather than through charities such as CRY. However, ECG screening is not sufficient to diagnose an individual with HCM.1
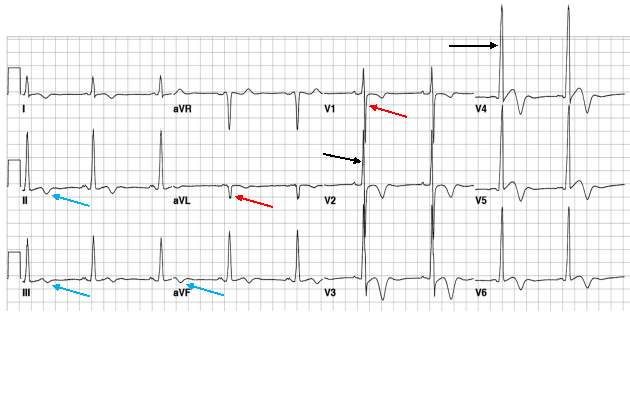
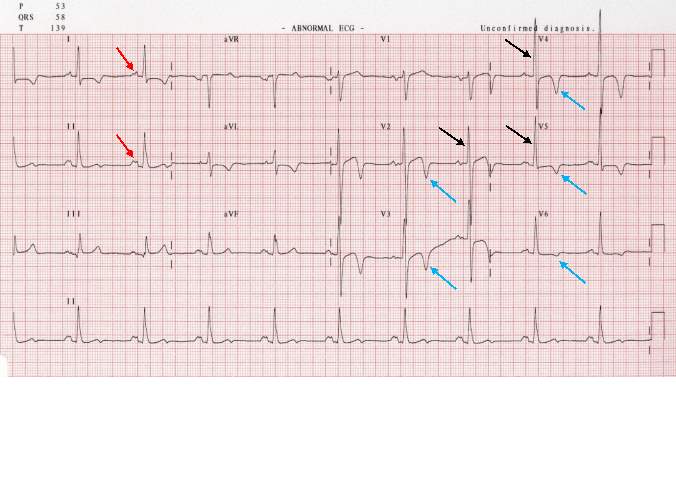
3.4 Screening using ECHO
ECHO uses sound waves to create an image of the heart, similar to how ultrasound scans are used in pregnant women. ECHO builds a detailed structure of the heart and valves, and gives information on the effectiveness of the systolic cycle of moving blood through the heart. ECHO can be used in any population, but is especially useful in screening neonates and young children due to the pain free, easy procedure.24 The standard ECHO, a transthoracic echocardiogram, involves lubricating jelly being rubbed on the chest and a probe which emits high frequency sound waves to the heart. The sound waves are then reflected off the cardiac structures and allows the probe to build an image of the heart. Transoesophageal ECHO is more usually used to build a closer and more defined image of the heart valves, but it is more invasive than the transthoracic variation and is also more expensive. Transthoracic ECHO provides another cheap, simple and easy screening procedure which could be readily available on a large scale.24 ECHO screening is usually done after recommendation from ECG screening, and is used to completely confirm a diagnosis of HCM.
3.5 Screening using cMRI
Cardiovascular magnetic resonance imaging (cMRI) is another non-invasive medical imaging technology.25 cMRI can be used in conjunction with ECG and ECHO when these methods do not provide enough evidence to allow a diagnosis to be made, such as when hypertrophy occurs in areas in the heart which cannot be visualised adequately by ECHO. cMRI can be used to diagnose a vast range of heart conditions including inherited heart conditions such as HCM.25 The test produces high resolution images which allow very accurate measurements to be made. Like ECG and ECHO it is also completely safe. Gadolinium is injected into the bloodstream, which provides contrast on the images, allowing detailed reconstructions to be made.25 Although cMRI usage is increasing, and most UK hospitals have access to cMRI with the rate increasing by approximately 15-20% each year, cMRI remains a much more expensive diagnostic test than either ECG or ECHO.25
3.6 Treatment and management
There is currently not a cure for HCM, and so treatments are used to manage the symptoms of the condition. Asymptomatic individuals should have a normal life expectancy, given that they avoid vigorous physical exercise and competitive sporting activities. Symptomatic individuals can take medications to control them. First-line treatments are beta-blockers which will work to relieve the contractile force of the cardiac muscle, reducing the heart rate, blood pressure and decreasing the chance of ectopic beats.26 Reducing the heart rate allows more time for passive ventricular filling.26 They can be used in individuals with and without outflow obstruction.1 Beta-blockers decrease the LVOT gradient, the cardiac workload and oxygen demand. Beta-blockers are also believed to reduce ventricular stiffness, believed to be caused by the sympatholytic (nerve impulse transmission inhibition) effects of the beta-blockade.1 Patients are usually put on a dose of 25mg metoprolol tartrate (Lopressor) twice daily, or metoprolol succinate (Toprol-XL) 50mg daily to alleviate symptoms to a goal of 60 beats per minute.26 If symptoms continue the dose can be increased by 25mg every few weeks, up to a maximum dose of 400mg/day.26 Beta-blockers side effects include: diarrhoea, stomach cramps, nausea, vomiting, rash, blurred vision, disorientation, insomnia, alopecia, muscle fatigue/cramps and various CNS effects e.g. headache & dizziness.26
Second-line treatments are non-dihydropyridine calcium-channel blockers such as Verapamil. These have a negative chronotropic effect, meaning the reduction of the heart rate, which increases diastolic relaxation time and preload, the end diastolic volume which stretches the heart to its largest dimensions.26 This works similarly to beta-blockers, in that it allows increased time for passive diastolic ventricular filling, and reduced outflow gradient.1 Usual dosage starts at 240mg daily, but can be increased every few weeks by 60mg up to a maximum dose of 460mg/day.26 Verapamil’s side effects include: dizziness, bradycardia, constipation, nausea, headache, tiredness, skin rash/itching, skin flushes, swelled ankles, weight gain, fainting, fever, dark urine and jaundice.26
Disopyramide, a Vaughan-Williams class IA antiarrhythmic drug, can also be used due to its negative inotropic effects that lead to improved diastolic function and decrease LVOT gradient.26 It has been proven to improve the outflow gradient of patients at rest, however it has significantly more severe side effect profile compared to beta-blockers or calcium-channel blockers.1 However despite this, under close professional supervision, it remains a valid and practical pharmacological treatment option for individuals who have continued to be symptomatic, with high outflow gradients, when beta-blockers and/or calcium-channel blockers have been unsuccessful.1 Disopyramide has no effect in patients without an outflow obstruction in HCM patients.1
Atrial fibrillation is a common symptom of HCM and is the most common arrhythmia of HCM, occurring in 20-30% of cases.1 Normal sinus rhythm can be restored by using direct current cardioversion if the patient is haemodynamically unstable, a procedure involving applying electrical currents to the heart through electrodes on the chest, or antiarrhythmic agents in stable patients.1 In stable patients with atrial fibrillation, the management options focus on 2 goals: rate versus rhythm control, and mitigating the risk of thromboembolism.1 Patient symptoms and treatment preferences are factors taken into account when deciding on which management option to take.1 Both the European Society of Cardiology and the American College of Cardiology/American Heart Association guidelines recommend that all HCM patients be treated with oral anticoagulation, or antiplatelet agents for individuals unable to take oral anticoagulation.1
Lifestyle choices such as the cessation of competitive sport or excessive exercise are recommended reduce the risk of a SCD incident and reduce the symptoms of HCM.
3.7 Surgical interventions
Patients suffering from the symptoms of heart failure with an outflow gradient ≥ 50mmHg, septal myectomy is the most effective treatment, and has been recommended by the American Heart Association guidelines.23 Septal myectomy involves removing part of the septum, allowing blood to flow freely by removing the outflow obstruction, and also relieving the symptoms of heart failure.13 Multiple studies have shown that permanently removing the thickened septal wall, and thus the obstruction, can relieve the symptoms of heart failure, lead to greater quality of life, and increase life expectancy to that similar to the general population.26
Percutaneous alcohol septal ablation also available, as a less invasive catheterised procedure. This procedure involves creating a small, controlled infarct in the cardiac tissue induced by pure alcohol infused into a cardiac artery.26 This causes some cardiac tissue to die, and subsequently leads to a reduction in cardiac tissue, leading to the obstruction being cleared and allowing blood flow through the heart to return to normal.
Internal cardioverter defibrillators can used as secondary prevention.27 They can be implanted in patients with HCM who have survived either ventricular tachycardia arrest or ventricular fibrillation.27 Internal cardioverter defibrillators are placed under the skin while the patient is under local anaesthetic. Small wires are attached to certain regions of the heart to detect the cardiac rhythm. If a dangerous rhythm is detected, the device will administer either pacing impulses to try correct the rhythm of the heart, small cardioversion shocks to restore normal heart rhythm, or large defibrillation shocks to restore normal heart rhythm. This constant correction of cardiac rhythm reduces the chance of arrhythmias from occurring, and if they do occur the defibrillator works to prevent the arrhythmia becoming fatal and restore the safe normal cardiac rhythm.
3.8 Other common conditions
Although hypertrophic myopathy is the most commonly inherited genetic cardiac disorder, there are many other conditions which exhibit similar ECG characteristics. Long QT syndrome (LQTS) is a condition which affects cardiac repolarization after a complete systolic cycle.26 This prolonged repolarization results in an extended QT interval which increase the risk of an irregular heartbeat, which can result in fainting, seizures or heart palpitations.26 The estimated prevalence is 1 in 2,000 individuals.26 Although it is commonly inherited, LQTS can be induced by some medical treatments including antibiotics, antihistamines, antidepressants, antipsychotics, diuretics and common cardiac medications.26 LQTS is one of the other major conditions associated with SCD, but can be easily diagnosed using ECG screening, and treated successfully with beta-blockers, implantable pacemakers or implantable cardioverter defibrillators if the symptoms are frequent or severe.23 Individuals diagnosed with LQTS can be advised not to play competitive sports or complete strenuous exercise. In certain situations individuals can be advised to remove startling noises which may cause stressful situations, such as alarm clocks, as stress can induce the associated symptoms of LQTS.26
Wolff-Parkinson-White syndrome (WPW) is another relatively common cardiac condition affecting approximately 1-3 in 1000 individuals according to NHS figures. WPW is a congenital condition although rarely presents until adolescence.28 The majority of cases are diagnosed after 20-40 years of age. WPW generally presents as supraventricular tachycardia, a symptom where the heart rate rapidly increases before slowing abruptly.28 During episodes of supraventricular tachycardia, individuals can experience heart palpitations, light-headedness, dyspnoea, chest pain, sweating, anxiousness, and fainting.28 The cause of WPW is associated with an accessory electrical connection between the atrium and ventricle, causing electrical bypass of the system, commonly in a bypass tract called a “Kent bundle” which is an embryological remnant.28 This allows tachycardia circuits to be established, where electrical signals from the sinoatrial node are continuously transmitted around the heart, causing continuous cardiac contraction.28 WPW can easily be diagnosed by ECG, patients will be asked to carry a portable ECG so the cardiac rhythm can be recorded during an episode. Treatments for WPW are mainly focussed on lifestyle changes, and training methods to calm yourself in episodes.28 Vagal manoeuvres (slowing heart by stimulating the vagus nerve), antiarrhythmic agents, and cardioversion are all treatments aimed at stopping an episode once it has begun.28 Lifestyle changes, antiarrhythmic agents and catheter ablation are treatments aimed at preventing future episodes.28
Commotio cordis is another common cause of SCD, however it is not associated with cardiac or genetic abnormalities. This condition is caused by a sudden blow to the precordial region, directly above the heart.29 If the blow occurs just as the cardiac T wave is starting, then the cardiac rhythm can be disrupted and the heart can go into cardiac arrest.29 This condition is common in certain sports, with hard solid balls such as baseball in the USA or cricket and hockey in the UK. Due to the nature of this condition the only way to prevent it from occurring is to introduce safety regulations into sports which have high risk. Regulations such as protective gear for individuals at risk, such as batters in cricket or goalkeepers in hockey, would reduce the incidence of this condition.
Another condition which commonly gets mistaken for HCM is a condition called athletes heart.22 In this condition the myocardium is enlarged as a natural physiological response to intensive physical exercise, which increases stroke volume and cardiac output. This hypertrophied cardiac muscle is not equivalent to HCM as it has no genetic causes and can occur in any individual who does intense aerobic training. It is also a non-lethal condition and poses no risk to athletes of suffering a SCD event. It can be mistaken for HCM because it appears to be physiologically identical to the pathological condition, due to similar hypertrophied ventricles.
4 Methods
The search engines PubMed and Cochrane Library were used and extensively searched to identify systematic reviews that outlined sudden cardiac death, hypertrophic cardiomyopathy and cardiac screening. For the PubMed the search terms used are shown in table 2. For the Cochrane Library search I used the search term: sudden cardiac death.
Inclusion criteria for this overview included:
- Reviews must report on ECG screening and HCM
- Reviews must investigate young adults or children
- Reviews must be in the English language
- There was no restriction on publication date to ensure maximum number of reviews could be included
From these searches 49 systematic reviews were identified to be potentially included. After reviewing and screening the titles and abstracts, 17 were excluded due to not being directly related to the topic. Remaining systematic reviews then had full text examination for eligibility. Studies not in the English language were excluded. Studies which did not analyse the effectiveness of ECG screening were excluded. Studies not related to HCM were excluded. This process is shown in figure 7. Although 1 review (Rodday et al) included individuals up to an age of 87 years old, the data from the rest of the review is exemplary and has therefore been included.
5 Results
The three systematic reviews which were included for overview looked at the effectiveness of ECG and ECHO as diagnostic tools.
5.1 Is screening reliable and accurate?
Rodday et al. provided a systematic overview of a number of cardiac conditions, but only the data regarding HCM has been selected.30 This systematic review included children and young adults in the age range of 3 to 87 years old, however the vast majority of the study population fell into the age group
Harmon et al. reviewed an ethnically and racially diverse group of 47, 137 athletes over 15 studies from Algeria, Spain, France, Germany, Greece, Switzerland, Qatar, the Netherlands, and the United Kingdom.31 This included athletes ranging from 5 to 39 years old. This review demonstrated that the sensitivity of ECG screening ranged from 79% to 98%, with the mean sensitivity being 94%. It showed that the sensitivity of ECG screening was consistently higher than both family history (20%) and physical examination (9%) and thus was the most accurate of the 3 screening tests. The specificity of ECG ranged from 90% to 96%, with the mean being 93%. This specificity was similar to both family history (94%) and physical examination (97%). ECG was found to have a positive likelihood ratio of 14.8, showing that a positive ECG test greatly increases the likelihood of disease. Out of 47, 137 athletes screened, 160 potentially lethal cardiovascular conditions were identified, or 1 in 294 athletes.
Calore et al. reviewed published studies related to the prevalence of the changes associated with HCM which show on ECG screening.32 Pooling of the data shows that of 2,171 individuals screened for cardiac conditions, 514 had some kind of ECG abnormality, which could be attributed to any of the potentially lethal cardiac conditions mentioned in the background. This review found that ECG abnormalities are observed in >90% of HCM patients. Table 3 shows data extrapolated from all 3 systematic reviews, regarding the reported relationship between the number of HCM cases found and the percentage of those which displayed an abnormality on an ECG scan. Pooled data shows that of the studies which reported on prevalence of HCM and ECG abnormalities, 526 cases of HCM were identified, and of those, 496 (94.3%) cases displayed single or multiple ECG abnormalities.
5.2 Does screening reduce the rate of mortality of SCD?
Table 4 shows combined data from all 3 systematic reviews regarding the number of individuals screened, and the number of HCM cases which were found as a result of the screening programmes. Pooling of the data shows that of 108,560 individuals screened by pre-participation programmes or screening of the general population, 130 cases of HCM were found and diagnosed. This equates to a rate of ~ 120 diagnosed per 100,000 screened. Assuming that a high proportion, if not all of these detected cases can be asymptomatic, this represents a substantial decrease in mortality risk from SCD.
6 Discussion
6.1 Screening as an effective diagnostic tool
Screening programmes have been shown to be able to successfully diagnose HCM and various other conditions associated with SCD. This overview has shown that screening programmes utilising ECG and ECHO are both highly sensitive and specific when diagnosing HCM and other potentially lethal cardiac disorders. The review conducted by Rodday et al. showed that ECG has a NPV close to 100%, but a PPV which was very low in every study. This could be due to the fact that this figure is the PPV for HCM, and does not take into account the other cardiac conditions mentioned previously, LQTS and WPW syndrome, which share similar abnormalities on ECG traces. Both LQTS and WPW syndrome demonstrate T wave inversions and large precordial voltages, which are both also associated with HCM. This means that the number of ECG abnormalities and the number of HCM cases in large screening programmes can be very varied. This also explains the great differences in false positives and negatives across the studies investigated. The exceptionally high NPV indicates that ECG abnormalities are extremely characteristic of cardiac defects and that if an ECG trace is normal, there is close to 0% chance of HCM or other cardiac conditions being present.
This could be used to fuel an argument that ECG screening is ineffective for the diagnosis of HCM, but as the data in table 3 shows 94.3% of HCM cases overviewed by the 3 reviews demonstrated some kind of ECG abnormality. This supports what was shown in Harmon et al. which found that ECG sensitivity was 94%. These findings indicate that ECG screening remains an effective tool for the diagnosis of HCM, but that there can often be need for further testing via ECHO or cMRI in order to give dangerous, abnormal hearts their correct diagnosis. The justification of providing ECG screening for HCM is also supported by the data found in the Calore et al. review which found that >90% of patients with a confirmed diagnosis of HCM also presented with an ECG abnormality representative of a cardiac condition. The results of this overview provide evidence supporting the use of ECG and, if required, ECHO screening for the identification of HCM and various other potentially fatal cardiac conditions.
This leads onto the suggestion that screening programmes will reduce the rate of mortality from SCD. The screening programmes have been shown to be effective in their ability to diagnose conditions that can potentially lead onto SCD. A large proportion of individuals who are diagnosed with HCM will be asymptomatic and as such may never have known that they had a condition associated with SCD. After diagnosis by the screening programmes asymptomatic individuals can begin to change their lifestyles, mainly by ceasing to partake in competitive sports or vigorous exercise, which can induce a SCD event. These individuals would have had no way of knowing they had a potentially lethal cardiac condition, other than potential relatives who were known to have the disease, and so were consistently competing in intense physical activity and consistently putting themselves at risk of a SCD event. Lifestyle changes will reduce the likelihood of death from SCD in these individuals, hence lowering the rate of mortality. It is unlikely that symptomatic individuals will have much effect of the rate of mortality due to lifestyle changes as they are likely to have already been to see medical professionals and previously been diagnosed, given treatments to manage their symptoms and have be taking necessary steps to change their lifestyle. Again however, because of these reasons it is unlikely that they would need to be screened in these screening programmes in the first place, as they are likely to already have a diagnosis. Symptomatic individuals with a diagnosis could be filtered out of the screening process, by using a pre-screening questionnaire and move immediately past this preliminary stage, straight onto treatment options including medications and surgery.
The estimated prevalence of SCD has been reported to be approximately 1 in 500.1 However, the data shown in table 2 shows that HCM was present in 130 out 108,560. This leads to an approximate prevalence level of 1 in 835 individuals. The prevalence found in this overview is 1.67 times less than the prevalence reported by previous studies and NHS figures.1 However, this prevalence only includes those cases which have been identified by screening programmes. Many cases of HCM will go undetected as individuals may never suffer from symptoms of the disease, and continue to live life without ever knowing they have HCM, unless they have been identified by ECG or ECHO screening. These figures could lead researchers to believe that the actual prevalence of HCM is much lower than has been previously reported. More studies are required to further investigate the prevalence of HCM to establish if this lower prevalence rate than reported is accurate.
It is believed that the case-fatality rate of HCM is approximately 60 per 100,000 in England and Wales.33 The data demonstrated in table 4 shows that in the screening programmes included in this overview, the rate of individuals diagnosed with HCM is approximately 120 per 100,000. Although this is below the predicted prevalence in the general population of 200 per 100,000 it still represents a fairly large cohort of individuals with HCM, equating to approximately 40,000 individuals according to the UK census figures of the population below 40 years old (total population
All but 1 of the screening programmes included in this study were conducted outside of the UK. This leads on to the question as to whether the results of this overview would be representative of a UK population. 14 of the 18 studies taken from the 3 reviews for investigation were conducted either in North America or Europe, in majority white Caucasian individuals. Because of this, it can be assumed that these studies can be applied to the UK population, and that the incidence and prevalence of SCD are not significantly different in the UK. However, due to the nature of the UK being a very multicultural nation more investigation specifically in the UK would be needed to confidently establish the effectiveness of ECG in the UK population, considering there are large non-white Caucasian communities where the same genetic risk factors may not be applicable.
6.2 Strengths and limitations
The main strength of this overview is the fact that there has been a relatively large population size of 107,596 individuals screened, and it has combined the data relating to the number of individuals screened versus the number of HCM cases identified. This large study population allows the calculated mean to be more precise and narrower margin of error. It also allows easier identification of outliers. However, there were no outliers found in this overview.
Another strength of this overview was the nature of the search strategy. This overview utilised a comprehensive search of multiple search engines. This, combined with the broad search terms and associated synonyms, meant that as many studies which were eligible for inclusion as possible were present in the initial search, before any screening and full text examination took place to exclude studies. This allows the findings of this overview to be more reliable.
There are several limitations to this overview. Firstly, in this overview the literature search was limited to Pubmed and Cochrane Library searches. Even though these two search engines index most biomedical articles, meaning it is doubtful that relevant important systematic reviews have not been included, there is still the chance that some important systematic reviews were missed by this search structure. It is possible that some publication bias may have taken place as it could be believed that studies which indicate ECG and ECHO screening as an unsuccessful measure of diagnosis or accuracy will not have been published. If these studies exist and have not been published then that would inflate the results of this overview. There is also the possibility of bias due to the fact that 1 review (Calore et al.) made no mention or comment on their screening for bias. It also made no statement regarding whether the studies included in the review were randomised or how they were controlled.
There is a high level of heterogeneity between the studies included in the 3 systematic reviews overviewed. Populations vary by age, race, gender and perhaps most significantly different diagnostic criteria and screening approaches. Some studies have used the European Society of Cardiology (ESC) guidelines published in 2015, ESC 2010 guidelines, ESC 2005 guidelines, and some have used the American College of Cardiology/American Heart Association (ACC/AHA) Guidelines published in 2014. Although there is relatively little difference between the two sets of guidelines, there should be a review of the number of HCM cases found via the ESC guidelines versus the ACC/AHA guidelines to identify if there is any significant difference between the screening guidelines, to reduce the chance of misclassification bias. Also the majority of the study population where white males and thus more studies are required to further investigate the effectiveness of screening programmes in other ethnicities and women.
7 Conclusion
SCD has remained a large risk for young adults (below 40 years old) and children. The condition with the highest death rate from SCD is HCM, and it remains a fairly common condition in approximately 1 in 500 individuals in the UK according to figures published by the NHS, however this overview shows that this rate is perhaps incorrect and the actual prevalence of HCM is much lower. The mortality rate of HCM has been recorded to be approximately 6 deaths per 10,000, and HCM remains the largest underlying cause of all SCD events. This overview has shown that screening programmes involving ECG and/or ECHO technology can be used to accurately detect and diagnose cases of HCM and consequently prevent SCD events. ECG screening was shown to have a sensitivity of 94% and a specificity of 93%, and that ECG abnormalities were found in 94.3% of HCM cases. These results show that ECG screening is an effective method for the detection and diagnosis of HCM in both symptomatic and asymptomatic individuals. There was however a large number of false-positives and false-negatives associated with the screening programmes, but this can be predicted as being due to the fact that many other potentially lethal cardiac conditions, such as LQTS and WPW syndrome, share similar ECG abnormalities on traces as HCM such as large precordial voltages and deep “dagger” like Q waves.
Due to these findings, it can be recommended that screening programmes involving ECG and ECHO should be provided by the NHS. Not on a large scale but only in the demographics most at risk of suffering a SCD event, mainly young athletes and children below the age of 40. If implemented into this demographic, screening programmes have the potential to detect potentially lethal cardiac conditions, and allow healthcare professionals to recommend the appropriate treatments and lifestyle modifications which will reduce the risk of SCD events occurring. Due to the nature of ECG screening, it is hard to say that the false-positives are detrimental to the recommendation of screening programmes becoming readily available, due to the fact that even though HCM may not be detected, as was the focus of this overview, other dangerous cardiac conditions will be found which is beneficial to the general population’s health. These factors are currently the reasons that charities in the UK such as CRY offer these services, and there is argument that the NHS should take more responsibility for individuals at risk of SCD, to ensure the safety of at risk British citizens. There is clear need for review in the NHS regarding their conceived perceptions of the danger SCD and the conditions associated with it such as HCM pose to the general public, especially to young adult and child athletes. If possible the NHS should provide screening programmes for young athletes, potentially done at a certain age during a child’s school career. This could be done fairly cheaply considering the approximate cost per patient per electrocardiogram is £8.66 according to the NICE figures on preoperative screening costs.34 This will allow screening programmes to identify dangerous, potentially lethal, asymptomatic cardiac conditions such as HCM, to prevent the unnecessary, treatable deaths from SCD of occurring.
9 References
1. Houston BA, Stevens GR. Hypertrophic Cardiomyopathy: A Review. Clin Med Insights Cardiol. 2014; 8(Suppl 1): 53-65.
2. Zipes D, Wellens H. Sudden Cardiac Death. Circulation. 1998; 98(21): 2334-51.
3. UK National Screening Committee. Screening for Sudden Cardiac Death. 19th March 2015.
4. NHS. Coronary Heart Disease: Chapter 8, Arrhythmias and Sudden Cardiac Death. Department of Health, March 2005.
5. Elliot PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014; 35(39): 2733-79.
6. Fifer M, Vlahakes G. Management of Symptoms in Hypertrophic Cardiomyopathy. Circulation. 2008; 117: 429-39.
7. Frenneaux MP, Counihan PJ, Caforio AL, Chikamori T, McKenna WJ. Abnormal blood pressure response during exercise in hypertrophic cardiomyopathy. Circulation. 1990; 82: 1995–2002.
8. Schwammenthal E, Nakatani S, He S, Hopmeyer J, Sagie A, Weyman A, et al. Mechanism of Mitral Regurgitation in Hypertrophic Cardiomyopathy. Circulation. 1998; 98: 856-65.
9. Gwathmey JK, Warren SE, Briggs GM, Copelas L, Feldman MD, Philips PJ, et al. Diastolic dysfunction in hypertrophic cardiomyopathy. Effect on active force generation during systole. J Clin Invest. 1991; 87(3): 1023-31.
10. Rosmini S, Biagini E, O’Mahony C, Bulluck H, Ruozi N, Lopes L, et al. Relationship between aetiology and left ventricular systolic dysfunction in hypertrophic cardiomyopathy. Heart. 2017; 103: 300-6.
11. Elliot PM, Kaski JC, Prasad K, Seo H, Slade AK, Goldman JH, et al. Chest pain during daily life in patients with hypertrophic cardiomyopathy: an ambulatory electrocardiographic study. Eur Heart J. 1996; 17(7): 1056-64.
12. Kuck K. Arrhythmias in Hypertrophic Cardiomyopathy. PACE. 2006; 20: 2706-13.
13. Pantazis A, Vischer AS, Perez-Tome MC, Castelletti S. Diagnosis and management of hypertrophic cardiomyopathy. Echo Res Pract. 2015; 2(1): 45-53.
14. Ellims AH. Hypertrophic cardiomyopathy in the adolescent. Aust Fam Physician. 2017; 46 (8): 553-7.
15. Marsiglia JDC, Pereira AC. Hypertrophic Cardiomyopathy: How to Mutations Lead to Disease? Arq Bras Cardiol. 2014; 102(3): 295-304.
16. Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013; 381(9862): 242-55.
17. Lopez LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, Dalageorgou C, et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet. 2013; 50: 228-39.
18. Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008;19(1):104–10.
19. Landstrom AP, Ackerman MJ. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation. 2010;122(23):2441–9.
20. Schmehil C, Malhotra D, Patel DR. Cardiac screening to prevent sudden death in young athletes. Transl Pediatr. 2017; 6(3): 199-206.
21. Bacharova L, Ugander M. Left ventricular hypertrophy: The relationship between the electrocardiogram and cardiovascular magnetic resonance imaging. Ann Noninvasive Electrocardiol. 2014; 19(6): 524-33.
22. Corrado D, Basso C, Parvei A, Michieli P, Schiavon M, Thiene G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006; 296(13): 1593-601.
23. Maron BJ, Thompson PD, Ackerman MJ, Balady G, Berger S, Cohen D, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2007; 115(12): 1643-455
24. Silvestry FE, Kerber RE, Brook MM, Carroll JD, Eberman KM, Goldstein SA, et al. Echocardiography-guided interventions. J Am Soc Echocardiogr. 2009; 22(3): 213-31.
25. Villeundas R, Kadish AH. Cardiac magnetic resonance for risk stratification: the sudden death risk portrayed. Prog Cardiovasc Dis. 2008; 51(2): 128-34.
26. Mizusawa Y, Horie M, Wilde AA. Genetics and clinical advances in congenital long QT syndrome. Circ J. 2014; 78(12): 2827-33.
27. Maron BJ, Spirito P, Ackerman MJ, Casey SA, Semsarian C, Estes NA, et al. Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J AM Coll Cardiol. 2013; 61(14): 1527-35.
28. Hollman A. The Wolff-Parkinson-White syndrome: a very long follow-up. Am J Cardiol. 2014; 113(10): 1751-2.
29. Maron BJ, Estes NA. Commotio Cordis. N Eng J Med. 2010; 362(10): 917-27.
30. Rodday AM, Triedman JK, Alexander ME, Cohen JT, Ip S, Newburger JW, et al. Electrocardiogram screening for disorders that cause sudden cardiac death in asymptomatic children: a meta-analysis. Paediatrics. 2012; 129(4): 999-1010.
31. Harmon KG, Zigman M, Drezner JA. The effectiveness of screening history, physical exam, and ECG to detect potentially lethal cardiac disorders in athletes: A systematic review/meta-analysis. J Electrocardiol. 2015; 48(3): 329-38.
32. Calore C, Zorzi A, Corrado D. Clinical meaning of isolated increase of QRS voltages in hypertrophic cardiomyopathy versus athlete’s heart. J Electrocardiol. 2015; 48(3): 373-9.
33. Wald DS, Law M, Morris JK. Mortality from hypertrophic cardiomyopathy in England and Wales: clinical and screening implications. Int J Cardiol. 2004; 97(3): 479-84.
34. National Institute for Health and Care Excellence. Preoperative tests: Routine preoperative tests for elective surgery. October 2015. Page 9.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Cardiology"
Cardiology is a medical speciality that deals with diseases, the function, and defects of the heart and cardiovascular system such as valvular heart disease, coronary heart disease, heart failure and others. Cardiologists diagnose and treat patients with such conditions.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: