Microglial Activation and Cancer Induced Bone Pain: Literature Review
Info: 2856 words (11 pages) Example Literature Review
Published: 26th Jan 2022
Tagged: CancerBiomedical Science
Microglial Development
Microglia are the resident macrophage-like cells of the central nervous system (CNS) and are the primary innate immune responders in a defense network that covers the entire brain parenchyma (Kettenmann, Hanisch, Noda, & Verkhratsky, 2011). They colonize the CNS during embryonic development from bone marrow derived hematopoietic stem cells that originate in the yolk sac and further proliferate to give rise to fully matured microglia (Alliot, Godin, & Pessac, 1999). This initial population of microglia within the CNS is referred to as resident microglia and they continue to invade the CNS during embryonic development until the blood brain barrier (BBB) is formed, after which they are never replenished (Milligan & Watkins, 2009; Matcovitch-Natan et al., 2016). Notably, the development of resident microglia is dependent upon the secreted cytokine, colony-stimulating factor 1, as it is known to regulate the proliferation, differentiation, and survival of hematopoietic stem cells (Elmore et al., 2014).
Following the formation of the BBB, self-renewal is the only source of new microglia in the healthy CNS (Matcovitch-Natan et al., 2016). These cells are referred to as perivascular microglia and are continually replenished in adulthood by bone marrow derived hematopoietic precursors (Milligan & Watkins, 2009). Although it remains a question of debate whether immune cells can cross the BBB, there is increasing evidence that bone marrow derived cells are capable of entering the CNS and differentiating into microglia in adults (Lull & Block, 2010). This has been shown to be possible even when the BBB remains intact, suggesting a mechanism for entry into the CNS. However, the mechanisms through which circulating cells are recruited to the CNS, and whether they enter the CNS under normal, resting conditions, are poorly understood (Lull & Block, 2010).
Resting Microglia and Microglial Activation
Phenotypically, in a healthy CNS, microglia are characterized by a ramified morphology consisting of small cell bodies with numerous slender branching processes. This is termed “resting microglia” and the immunological phenotype of this state is characterized by the low expression of major histocompatibility complex (MHC) proteins and other antigen-presenting surface receptors (Lull & Block, 2010). Despite being in a quiescent state, resting microglia have highly motile processes and are continually surveying the health of surrounding cells by constantly extending and retracting their processes, looking for changes in their microenvironment. Consequently, microglia have extensive signaling pathways whereby they can respond to extracellular signals, communicate with other cells including neurons and cells of the immune system, and maintain homeostasis (Kettenmann et al., 2011).
As microglia are considered to be the most susceptible sensors of brain pathology, the detection of any pathogens, signs of brain injury or nervous system dysfunction can cause microglia to undergo dramatic alterations in morphology (Kettenmann et al., 2011). This entails changing from “resting” ramified microglia into an amoeboid shape with short or nonexistent processes. This morphological change is also accompanied by changes in signaling and gene expression that result in changes in surface receptor expression, the release of proinflammatory factors such as tumor necrosis factor- (TNF-) α, Interleukin- (IL-) 1β, IL-12, reactive oxygen species (ROS), prostaglandins (PGs), chemokines and neurotoxic factors, among other substances such as glutamate (Lull & Block, 2010). The cumulative effect of these changes is a shift from “resting” to “activated” microglia.
Upon activation, microglia become phagocytic and seek to remove damaged cells and debris through phagocytosis (Kettenmann et al., 2011). The substances released by activated microglia further activate additional nearby astrocytes, microglia, and neurons to aid in this process, creating a positive feedback loop (Milligan & Watkins, 2009). Thus, once the harmful stimulus has been dealt with, it is imperative that the microglial inflammatory response be dampened and resolved. This is achieved by microglia releasing anti-inflammatory cytokines such as transforming growth factor-β, IL-4, and IL-10, and expression of Arginase-1(Lull & Block, 2010). These immunomodulatory mediators inhibit the release of proinflammatory factors from immune and nonimmune cells and promote tissue regeneration, thereby facilitating a return to homeostasis.
However, when the resolution phase of the inflammatory microglial response is altered, chronic microglial activation can ensue and results in a prolonged inflammatory response (Lull & Block, 2010). This can lead to tissue and neuronal damage and possibly cell death. It is for this reason that chronic microglial activation has ben implicated in the pathology of many neurodegenerative disorders such as Parkinson’s disease, Alzheimer’s disease, multiple sclerosis, and AIDS (Lull & Block, 2010).
Microglial Glutamate Release
The activation of microglia promotes the release of ROS, and thus creates a state of oxidative stress Is this intra- or extracellular disruption of redox homeostasis? (Lull & Block, 2010). This fosters the synthesis of glutathione (GSH), as it is an antioxidant that serves as a free radical scavenger to neutralize the harmful effects of oxidative stress and protects the activated microglia (Lewerenz et al., 2013).
GSH is synthesized in the cytosol through the enzymes γ-glutamylcysteine synthetase and GSH synthetase and involves cysteine as the rate-limiting substrate, which is imported into the cell using the system xc– amino acid antiporter. This antiporter mediates the 1:1 exchange of extracellular cystine, the oxidized form of cysteine, and intracellular glutamate across the cellular plasma membrane (Lewerenz et al., 2013).
Thus, when there is an increased need for the synthesis of GSH, as is the case when activated microglia release ROS into the extracellular environment?, microglia upregulate the expression of xCT, the functional subunit of the system xc– antiporter (Lewerenz et al., 2013). The overall effect of this is excess glutamate release, which alters glutamate homeostasis. As glutamate is a major excitatory neurotransmitter in the CNS, elevated levels of glutamate result in the over-activation of glutamate specific receptors such as the N-methyl-D-aspartate (NMDA) receptor and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor. This leads to a state of excitotoxicity and results in neuronal damage and death (Appendix Figure 1.) (Bridges, Natale, & Patel, 2012).
Cancer Induced Bone Pain
Many types of cancer, particularly breast, prostate, and lung cancers, are characterized by a strong propensity to metastasize to the bone (Lozano-Ondoua, Symons-Liguori, & Vanderah, 2013). These bone metastases have the potential to dramatically change the bone microenvironment, altering osteoclast and osteoblast functions, resulting in a range of pathological consequences such as skeletal remodeling, fractures, anemia, hypocalcaemia, and cancer-induced bone pain (CIBP) (Ungard, Seidlitz, & Singh, 2012).
CIBP is the most common symptom of bone metastasis, being present in roughly one third of patients with bone metastases, and can present as ongoing or breakthrough pain in any location within the bone (Lozano-Ondoua et al., 2013). Ongoing pain is the most common experience of CIBP and is described as dull in character, persistent, and increases in intensity over time, whereas breakthrough pain is often more intense and irregular and can be characterized as transient and debilitating exacerbations of bone pain. Breakthrough pain can ensue spontaneously, without any trigger event, or can be associated with the movement of the tumor bearing bone (Ungard et al., 2012). The cumulative effects of bone metastasis and the pain associated with it is a reduced quality of life and functional status for cancer patients.
CIBP has further been established as a unique pain state from other chronic pain states such as inflammatory or neuropathic pain, as it is characterized by unique neurochemical changes in the spinal cord and dorsal root ganglion (Honore et al., 2000). Although CIBP is considered to be a syndrome with a range of mechanisms contributing to the development and maintenance of pain, such as the alteration of osteoclast and osteoblast function by factors secreted by tumor cells, a notable contributor to CIBP is the release of glutamate (Lozano-Ondoua et al., 2013).
Glutamate is a tumor-derived substance that is secreted from cancer cells via the system xc– antiporter (Ungard et al., 2012). The expression of system xc– by cancer cells is thought to confer protection against oxidative stress as an adaptation to a metabolic abnormality known as the Warburg effect, the observation that cancer cells rely upon anaerobic glycolysis for ATP generation in low-glucose, hypoxic environments (Lozano-Ondoua et al., 2013). The Warburg effect results in the inefficient use of glucose and excessive generation of ROS as the normal byproducts of cellular respiration. As the major physiological role of system xc– is to support the synthesis of GSH to protect cell from damage by ROS, system xc– is critical to cancer cells as a mediator of cellular protection (Lozano-Ondoua et al., 2013). Also, aggressive cancer cells switch from glucose metabolism to relying on the utilization of glutamine as an energy source. Increased glutaminolysis via glutaminase activity leads to an increase in cellular glutamate levels, which fuel the Kreb’s cycle via conversion into alphaketoglutarate. However, it is thought that glutamate is produced in excess of what a cell requires for its metabolism.
However, the consequence of cystine uptake via system xc– is the obligatory secretion of glutamate into the extracellular space. In addition to serving as the predominant excitatory neurotransmitter in the CNS, glutamate is also a well-established algogenic substance (Ungard et al., 2012). Therefore, glutamate derived from cancer cells may evoke brain edema (how? Please reference this.), nociceptive signaling through chronic glutamate receptor activation, and excitotoxic cell death, leading to the persistent nociceptive states found in patients with CIBP. Furthermore, because glutamate is a vital intracellular signaling molecule in bone homeostasis and function, elevated glutamate has the potential to disrupt normal bone metabolism and signaling, contributing to the deregulation of bone remodeling and an overall increase in pain (Lozano-Ondoua et al., 2013);(Ungard et al., 2012).
Microglia and Cancer Induced Bone Pain
Treatment of CIBP is commonly done through adherence to the World Health Organization’s (WHOs) analgesic ladder, a stepwise progression from non-opioid analgesics to strong opioids (Kane, Hoskin, & Bennett, 2015). Adjuvant therapies such as radiotherapy, bisphosphonates, and corticosteroids can also be used.; However, it has been shown that at least 20-40% of cancer pain is not adequately relieved by the application of the WHO analgesic ladder (Weiss, Emanuel, Fairclough, & Emanuel, 2001). Additionally, limited success has been achieved at relieving pain long term, with more than 50% of patients experiencing pain that reoccurrs shortly after treatment (Kane et al., 2015). Therefore, the limited effectiveness of current pain control therapies proves to be an important area of research, and the investigation of novel mechanisms for the treatment of CIBP is not only of great clinical relevance but is critical to maintain a patient’s quality of life.
Previous literature has established the role of glutamate, among other mechanisms, in the propagation of CIBP (Lozano-Ondoua et al., 2013; Ungard et al., 2012). Distinct from the glutamate release of cancer cells is the release of glutamate by activated microglial cells (Lull & Block, 2010). As it has been determined that microglial activation can ensue in responses to immunological stimuli, it would be of interest to investigate whether CIBP and the sensitization of surrounding nociceptors might lead to the activation of microglia. This would further contribute to the release of excess glutamate and result in excitotoxicity via a distinct mechanism, exacerbating pain associated with CIBP.
Furthermore, tumor cells are known to secrete various substances such as TNF-α and IL-1β, which are substances that are known to activate microglia (Ungard et al., 2012). As such, cytokines secreted by cancer cells could be another contributor to the chronic activation of microglia and contribute to microglial glutamate release and the maintenance or propagation of CIBP.
Therefore, if a relationship between microglial glutamate release and CIBP is established, it could not only serve as a novel target for pain management strategies for CIBP, but the effects of existing therapeutic strategies such as the system xc– inhibitor, sulfasalazine, could be re-evaluated in the context of microglial glutamate release and the associated pain that may ensue.
Appendix
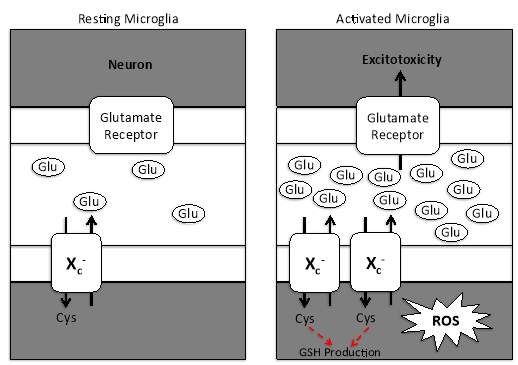
Figure 1. Microglial activation upregulates the expression of system xc– and contributes to increased glutamate release.
In response to pathological and physiological changes in the central nervous system, microglia undergo a complex activation process. This activation is accompanied by the release of reactive oxygen species, which creates a state of oxidative stress. Consequently, microglia upregulate the expression of system xc– and cysteine is imported into the cell for the synthesis of glutathione. The result is the increased secretion of glutamate into the extracellular space, which over-activates glutamate receptors, resulting in excitotoxicity. This image was adapted from Lewerenz, J. et al. (2013), 18(5), 522-555.
References
Alliot, F., Godin, I., & Pessac, B. (1999). Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Developmental Brain Research, 117(2), 145–152.
Bridges, R. J., Natale, N. R., & Patel, S. A. (2012). System xc-cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. British Journal of Pharmacology, 165(1), 20–34.
Elmore, M. R. P., Najafi, A. R., Koike, M. A., Dagher, N. N., Spangenberg, E. E., Rice, R. A., … others. (2014). CSF1 receptor signaling is necessary for microglia viability, which unmasks a cell that rapidly repopulates the microglia-depleted adult brain. Neuron, 82(2), 380.
Honore, P., Rogers, S. D., Schwei, M. J., Salak-Johnson, J. L., Luger, N. M., Sabino, M. C., … Mantyh, P. W. (2000). Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience, 98(3), 585–598.
Kane, C. M., Hoskin, P., & Bennett, M. I. (2015). Cancer induced bone pain. Bmj, 350, h315.
Kettenmann, H., Hanisch, U.-K., Noda, M., & Verkhratsky, A. (2011). Physiology of microglia. Physiological Reviews, 91(2), 461–553.
Lewerenz, J., Hewett, S. J., Huang, Y., Lambros, M., Gout, P. W., Kalivas, P. W., … others. (2013). The cystine/glutamate antiporter system xc- in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxidants & Redox Signaling, 18(5), 522–555.
Lozano-Ondoua, A. N., Symons-Liguori, A. M., & Vanderah, T. W. (2013). Mechanisms of cancer-induced bone pain. Neuroscience Letters, 557(0 0), 52.
Lull, M. E., & Block, M. L. (2010). Microglial activation and chronic neurodegeneration. Neurotherapeutics, 7(4), 354–365.
Matcovitch-Natan, O., Winter, D. R., Giladi, A., Aguilar, S. V., Spinrad, A., Sarrazin, S., … others. (2016). Microglia development follows a stepwise program to regulate brain homeostasis. Science, 353(6301), aad8670.
Milligan, E. D., & Watkins, L. R. (2009). Pathological and protective roles of glia in chronic pain. Nature Reviews. Neuroscience, 10(1), 23.
Ungard, R. G., Seidlitz, E. P., & Singh, G. (2012). Oxidative stress and cancer pain. Canadian Journal of Physiology and Pharmacology, 91(1), 31–37.
Weiss, S. C., Emanuel, L. L., Fairclough, D. L., & Emanuel, E. J. (2001). Understanding the experience of pain in terminally ill patients. The Lancet, 357(9265), 1311–1315.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biomedical Science"
Biomedical Science focuses on how cells, organs and systems function in the human body and underpins much of modern medicine. Biomedical Science applies parts of natural and/or formal sciences to help develop advances in healthcare.
Related Articles

DMCA / Removal Request
If you are the original writer of this literature review and no longer wish to have your work published on the UKDiss.com website then please: