Research Proposal for Fragmant Library for Future Drugs
Info: 4988 words (20 pages) Example Research Project
Published: 11th Jan 2022
SUMMARY OF THE THESIS PROJECT
Fragment-Based Lead Discovery (FBLD) is becoming a viable complement and alternative to traditional high-throughput screens for discovering the leads of future drugs. FBLD involves the screening of libraries of compound fragments to search for binders to target proteins. These binders can then be used as chemical biology probes, functional modulators or scaffolds to custom design quality lead compounds. FBLD is a validated approach which has led to compounds in the clinic/market. Thus, designing fragment libraries is crucial for FBLD.
Thesis work describes the series of development of various fragment libraries. We developed a new fragment library (INRS 1H) with a combination of chemoinformatics, and experimental NMR filters will be evaluated later, however, chemoinformatics based filters were employed to remove undesirable compounds (reactive, toxic, unstable, aggregators) and to prioritize desirable compounds (3D dimensionality, solubility, substructures). The diversity of the library is evaluated using Murcko scaffolds. Follow-up strategies will be implemented to detect compound aggregation and filter out promiscuous ligands. Smart pooling NMR-based screening methods will be introduced to identify the best binders to target proteins/RNA.
RESEARCH SUMMARY OF THE ARTICLE
In the drug discovery pipeline, when a researcher finds a hit from screening process, after testing for artifacts and gathering as much as structural information possible, the next process is usually to test the “analogs” or “structurally similar” compounds for improved potency. These analogs can come from two major sources:
a) Pharma companies generally have tremendous compound / fragment libraries that is easy enough for testing.
b) There are various commercial vendors, enabling “analog by catalog” search.
But, sorting through millions of compounds to find the suitable compound and order would be a tedious task. Thus, the researchers in Astex developed a computational tool “The Fragment Network” to rationalize the substructure search using a graph database; this tool allows the user to search through the “in-house” libraries as well as the commercial vendors, for ordering the compounds. The advantage of this tool is one can search through millions of compounds in a fraction of seconds.
In the fragment network tool, 4-hydroxy biphenyl is illustrated. Each fragment is computationally sliced into different components like hydroxyl or phenyl ring; edges are the connection between the components like carbon-carbon bonds. The fragment network tool has five million compounds up to 24 non-hydrogen atoms, but their fragment library has no more than 16 heavy atoms count. These searches are further annotated based on whether they are readily available from reliable vendors or from the in-house fragment library. The fragment network can group compounds by type – for example, the linker replacements are grouped separately from the ring substitutions. This grouping of the results, representing the areas of the less populated chemical-space.
The researchers applied the Fragment Network into their two previously reported campaigns against protein kinase B and HCV NS3, the program detected most changes examined by the medicinal chemists, and best fragments were not readily available and needed to be synthesized. The limitations of the program are,
a) The chemical structures are not visualized.
b) The tautomer is represented as a unique node; better representation of the tautomer would enhance the utility of the program.
c) The ring systems are linked if they share a common substituent. However, a set of additional edges to join similar ring systems would be beneficial.
d) Fragment Network could be modified, for the hits to lead and lead optimization stages, such as adding more relevant information such as NMR screening data, adding relevant binding site information.
Upgrading Fragment Network program will be highly beneficial not only for substructure searching but also for “in-house” data mining with the experimental data.
The authors had a set of “hits” from their screening campaign. This research proposal consists of “hits to lead” phase as a research proposal.
BACKGROUND
Drug discovery technologies are rapidly changing. Few technologies emerge, flourish and at one point in time, it challenges the existing technologies, some technologies quickly discontinued. Over the past two decades, fragment-based drug design (FBDD) has revolutionized the pharmaceutical industry with a series of compounds entering the clinic (Erlanson et al., 2016). Even though widely practiced in pharma, it is increasingly becoming popular in academia (Bulfer et al., 2015). The initial approach identifies the critical starting points, that is very small molecules termed as (“fragments” or “drug seeds“) these are weak binders, that are moderately half the size of conventional drugs. These typical fragments can evolve into larger compounds, either by linking or merging fragments or by growing the fragments (Erlanson, 2011) . The main constraints are the need for a “golden” method that can accurately detect weak binding and apply various strategies for evolving the fragments into larger lead compounds (Jhoti, 2008).
Drug discovery pipeline
Figure 1 depicts the drug discovery pipeline in various stages. It consists of significant steps: Libraries, Targets, and Screens.
A) The initial discovery stage determines a compound library with the suitable drug-like properties to move forward as a potential clinical candidate.
B) In the target phase, using biochemistry tools to generate quality target protein using bacteria or yeast. Other techniques such as target markers, various functional assays for binding, displacement and enzymatic activity are utilized.
C) The next phase is the screening phase using NMR one can identify the consensus binding, free-state properties and check for the aggregation.
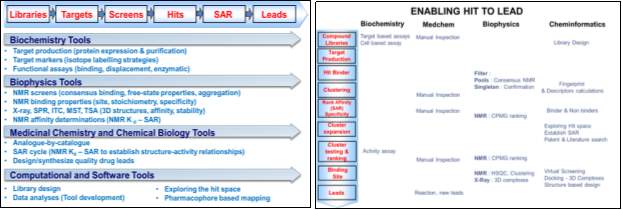
Figure 1:Drug discovery process. Figure 2: Enabling hit to lead phase.
In the hits to lead phase modifications to the hits to determining for optimization. The critical concept is which segments of the molecule are essential for biological activity and how it is modified, these are termed as Structure Activity Relationship (SAR), and only those tractable compounds are further chemically synthesized.
The work presented in this research proposal focuses on preclinical drug discovery, in particular, “hit to lead” phase specifically in the chemoinformatics domain. Figure 2, depicts the process of enabling hits to lead phase. The blue arrow indicates the various steps followed according to the different research domain (Biochem, Medchem, Biophysics, and Chemoinformatics). The major topics discussed will be on ligand-based drug discovery: Substructure based search: Virtual Screening, Clustering analysis using various fingerprints Pharmacophore-based analysis and Docking analysis.
Substructure based search: Virtual Screening
Virtual Screening (VS) of small molecules proposes a technique to enhance the drug discovery phase. Using VS, large databases of molecules (Example. ZINC database – http://zinc.docking.org/) can be computationally evaluated (Lavecchia et al., 2013). This allows the difficult and expensive experimental analysis to focus on the first fraction / phase of the screened library where the “active molecules” are expected to be enriched, thus saving time and resources (Shoichet, 2004). The virtual screening workflow begins by preparation of the database of small molecules for screening (Figure 3), adapted from (Andricopulo et al., 2008). Before starting the actual virtual screening, the database is usually refined to improve it with small molecules that have desirable properties, that is to eliminate the compounds with undesirable properties (Cramer et al., 1998). Different approaches used in combination with VS. First steps made with crude and computationally efficient methods and only the best scoring hit from the initial screens passing forward to stages, where more appropriate and accurate demanding methods are utilized (Cheng et al., 2012).
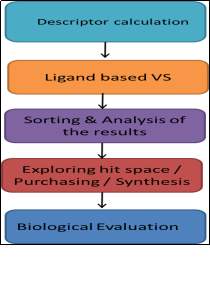
Ligand-based virtual screening methods utilize the idea of ‘similar topology principle’ that states the similar compounds should have similar biological activity. The similarity between the molecules is usually, evaluated with various types of molecules descriptors 1D, 2D, and 3D which depicts the properties of the molecules (Johnson et al., 1990). 1D descriptors calculated from the chemical formula of the molecules. 2D descriptors can generate substructure and connectivity information. 3D descriptors can provide a pharmacophore property, which additionally requires conformational information. In VS, the most commonly used methods are 2D and 3D descriptors. However, the 1D descriptor is not used alone in VS, but it is used as a filter criterion or in combination with 3D or 2D descriptor (Hong et al., 2008).
Clustering based analysis: Fingerprint and descriptor calculations
In 2016, (González-Medina et al., 2016) reviewed various fingerprint based descriptors., suggests the use of molecular scaffolds (Bemis and Murcko scaffolds), structural fingerprints (ECFP4, MACCS, FCFP4). Each representation has its own merits and demerits. Bemis and Murcko frameworks widely used for characterization of molecular scaffolds. Usually, the BM scaffolds capture the “cyclic skeletons,” which are part of the chemical structures but the side chain information not captured (Hu et al., 2011). ECFP4 fingerprints (Rogers et al., 2010) methodology used for capturing molecular features relevant to molecular activity; widely used in clustering, virtual screening and similarity searching. However, this fingerprint is harder to interpret as it captures the information of the whole structure. While, FCFP4, is functional class based fingerprint and it captures more abstract role-based substructural features. In 2017, a review by (Fernández-de Gortari et al., 2017) compares various molecular fingerprints for diversity analysis they used binary vectors Molecular ACCess System (MACCS) keys and database fingerprint (DFP). The performance of MACCS fingerprints was poor when compared to DFP fingerprint. Likewise, in 2016 (O’Boyle et al., 2016) compared structural fingerprints using a literature-based similarity, the authors compared the performance of MACCS keys with other fingerprints. Likely, MACCS compared perform poorest among other fingerprints.
Pharmacophore-based analysis
Pharmacophore-based fingerprints apply the concept of the atom pair fingerprints, that provides a simplified notation of significant interaction groups or atoms, not inevitably bonded to each other – any number of bonds can separate them. Otherwise, in pharmacophore fingerprints, three or four features are typically encoded and are termed as three and four-point pharmacophores. Further, pharmacophore fingerprints are used for calculating the protein-binding site similarity calculations (McGregor et al., 1999, McGregor et al., 2000).
In two-point pharmacophore fingerprints, there are two types: Typed Graph Distances (TGD) and Typed Atom Distances (TAD) these are considered an implementation of geometric atom pair and atom pair fingerprints, with (H-bond acceptor, H-bond donor, polar, hydrophobic, anion, cation). TGD employs graph based 2D representation of the molecules, whereas TGT utilizes 3D conformation. In three-point pharmacophore fingerprints, Typed Graph Triangles (TGT), Typed Atom Triangles (TAT), GpiDAPH3 and piDAPH3. The TGT and TAT employ triplets of atoms, whereas GpiDAPH3 uses graph-based and piDAPH3 uses unique 3D conformation and atoms with three properties (in the pi system, H-bond donor, H-bond acceptor and a total of eight possible combinations). In four-point pharmacophore fingerprints, piDAPH4 which employs the quartets of features and it is an extension of piDAPH3 (Chackalamannil et al., 2017, Koutsoukas et al., 2013).
HYPOTHESIS
Our project aims to deliver quality leads via the “hits to lead” phase. The title of our proposed project is “Identifying lead compounds from hits using chemoinformatics and SBDD approach”. The prime hypothesis for our project is identification of drug leads from the hits based on chemoinformatics and structure-based drug discovery.
General objective
As soon as one found hit from the screening campaign, the next step is to identify the similar compounds and look for the SAR, this is a challenging and most crucial step in the drug discovery, although the target structure of the protein may / may not be available in some cases.
Specific objectives
- Hit space exploration.
- Clustering based analysis.
- Pharmacophore-based mapping.
RESEARCH PLAN
Objective 1: Hit space exploration
Rationale
In the past few decades, discovering small molecule for treating various human disorders has been extraordinarily successful. The research in the pharmaceutical industries invests millions of dollars for the research community in search of novel treatments and medications. However, the success rate at the end stage failure is one of the most significant problems acquired by the giant pharma companies which remain expensive. VS are highly complementary for drug discovery campaigns. Virtual screening technologies appeared to be the most valuable and crucial tool for drug discovery during “hit to lead” phase. The aim of virtual screening is to run multiple algorithms, different methods, and choice of descriptors. It is eventually related to the “selection”/ “cherry picking” of individual compounds from a large database.
Proposed research
Goal: To identify substructure molecules in in-house and commercial libraries.
How: Based on virtual screening methodology.
Why is hit space exploration required?
Substructure search is very useful for identifying chemical structures that are similar, or analogs, in terms of the structural space they occupy.
This allows one to purchase or synthesize compounds to explore Structure-Activity Relationship (SAR) of chemical structures around a given molecular scaffold.
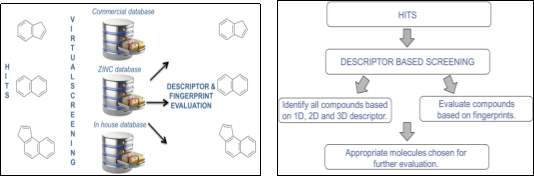
Figure 4: Outline of the proposed research and methodology
Methodology
Requirements:
- MOE 2018.01, Commercial, open-source databases: ZINC, chEMBL, Chemical Universe Database and in-house libraries.
- Certain number of experimental “hits” from drug discovery screening will be obtained.
- By employing “ligand-based virtual screening” methodology (Figure 4), using descriptor-based screening such as 2D, we will screen similar compounds using various databases including in-house libraries.
- Once we obtain the compounds, we will evaluate the compounds using various fingerprints such as MACCS, BIT_MACCS, ECFP1, 2, 4, 6, FCFP1, 2, 4, 6 and MPMFP). Similarity metrics will be calculated by Tanimoto Coefficient.
- Appropriate molecules are chosen for further evaluation.
- Note: All fingerprints have its own merits and demerits. So based on the performance of the fingerprints with the similarity search, the best performing fingerprints will be evaluated.
Expected results:
- In case of 50 hits from a screening campaign, there are possibilities to get at least a few thousands substructure based compounds.
Planned difficulties and an alternative plan:
- In case of million hits from the search, storing the data will be an alarming issue. Thus another server will be requested from Calcul Quebec or from Compute Canada.
Objective 2: Clustering based analysis
Rationale
Clustering techniques have been used in different areas of research geology, biology, computer science and drug discovery. Cluster analysis is the study of methods for grouping data quantitatively. Different algorithms are applied based on the data and the requirements. The clustering algorithm used in MOE 2018.01 is Jarvis-Patrick method. For example, Two molecules belong in the same cluster if they are similar to the same set of molecules. In case of few molecules, clustering is not required but when it is for a few thousands of data, grouping the molecular data based on the similarity metrics is important in drug discovery.
Proposed research
Goal: To cluster similar set of molecules based on the similarity of their fingerprint.
How: Based on the clustering analysis.
Why is the clustering search required?
- Compounds with similar structures/physical properties have similar activities.
- Similar compounds have at least one functional group in common.
- Similar compounds share the same frame/scaffold with various functional groups.
- Their descriptors or properties are similar.
- Their three-dimensional shapes are similar.
- Thus, clustering the similar compounds based on fingerprint will be more feasible.
Figure 5: Outline of the proposed research and methodology.
Methodology
Requirements:
- MOE 2018.01, results obtained from the substructure search, shape-based descriptors.
- A certain number of similar compounds from objective 1 will be obtained.
- By employing “clustering methodology,” (Figure 5) using shape-based screening such as x3D and i3D, we will cluster the similar compounds.
- Once we group the compounds, we will evaluate the compounds using various fingerprints such as PMI (Principal Moments of Inertia), EShape3D, BCUT 4D PEOE these descriptors are based on atomic charges, polarizabilities, H-bond donor, and acceptor abilities and H-bonding modes of intermolecular interaction. Similarity metrics will be calculated based on the Tanimoto Coefficient.
- Appropriate molecules are chosen for further evaluation.
Expected results:
- There are possibilities that we can cluster similar compounds using different fingerprints.
Planned difficulties and Alternative plan:
- Different clustering methodologies may be evaluated, but since MOE 2018.01 possess Jarvis-Patrick method. Clustering using different algorithms will be evaluated using KNIME workflow.
Objective 3: Pharmacophore-based analysis
Rationale
“A pharmacophore is defined as the interaction set of patterns of the ligands based on the 3D arrangements of pharmacophoric features (hydrogen bond donor, hydrogen bond acceptor, anionic, cationic) that define interaction types other than specific functional groups”. (Kaserer et al., 2015). The 3D structures of three or more ligands are aligned together and their common pharmacophore features are evaluated. Pharmacophore based methods are well established tools in drug discovery and specifically in the hit to lead phase.
Proposed research
Goal: To search for pharmacophoric features of the results obtained from the objective 2.
How: Based on pharmacophore mapping.
Why is pharmacophore search required?
- Smaller fragments may bind in various parts of the binding cavity.
- From the way a fragment docks, it may be difficult to discover how the ligand-containing the fragment may be docked.
- When the fragment is docked the different binding pose of the fragment in a ligand might not be a highest priority When fragments are merged and grown into a compound, the chances of finding the ligand pose seems better. Pharmacophores can direct the fragment to the location occupied when the fragments form a ligand.
Methodology
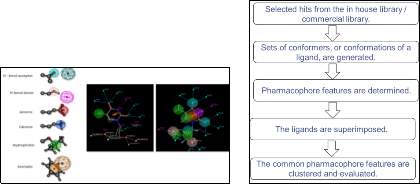
Figure 6: Outline of the proposed research and methodology.
Requirements:
- MOE 2018.01, results obtained from the shape based descriptors.
- A certain number of clustered similar compounds from objective 2 will be obtained.
- By employing “pharmacophore methodology,” (Figure 6) using conformers generate a set of compounds.
- Once we group the compounds, we will evaluate the compounds using various pharmacophoric fingerprints such as TAD, TAT, TGD, TGT, and GpiDAPH3: H-bond donor, H-bond acceptor, Anionic, Cationic, Hydrophobic and Aromatic features are calculated using these descriptors. Similarity metrics will be calculated based on the Tanimoto Coefficient.
- Appropriate molecules are chosen for further evaluation.
Expected results:
- The ligands are superimposed, and the common pharmacophoric features are clustered and evaluated.
Planned difficulties and Alternative Plan:
- Without the target structure of the protein, the ligand-based pharmacophore analysis is possible.
- However, the docking based analysis will provide more acceptable results.
Objective 3b: Binding site analysis
Rationale
The goal of molecular docking technique is to provide prediction of the target protein -ligand complex structure using computational tools. Molecular docking approach is a powerful mode to model the protein and small molecule interaction at the atomic level. This technique helps in characterizing the behavior of small molecules at their binding sites.
Proposed Research
Goal: To find the binding site analysis of the target protein and the ligand.
How: Based on docking analysis.
Why is binding site analysis required?
Molecular docking techniques are one of the most powerful tools for drug discovery. Precisely, binding site analysis play a vital role in identifying the lead compound. Because identifying and designing drug molecules are attributed to the protein binding site. Since, understanding and identifying the key interacting amino acids and its potential energies such as cation, ionic, electrostatic, steric interactions and other properties of the binding site permits to design novel lead series. Computational prediction of the binding site analysis saves research money and valuable time.
Methodology
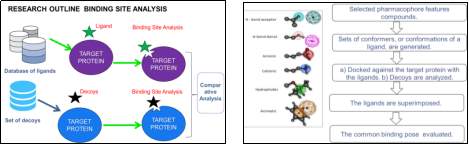
Figure 7: Outline of the proposed research and methodology.
Requirements:
MOE 2018.01, results obtained from the pharmacophore-based descriptors.
- Clustered and selected pharmacophore features compounds (Figure 7).
- Sets of conformers, or conformations of a ligand, are generated.
a) Docked against the target protein with the ligands.
b) Decoys are docked against the same target protein and analyzed.
- Decoys are set of useful ligands from this database http://dude.docking.org/subsets. DUD-E is developed to help standardize the molecular docking programs by utilizing challenging decoys. Decoys will be used for comparative analysis.
- Docked ligands are superimposed.
- The common binding pose evaluated.
Planned difficulties and an alternative plan:
- Only for some targets, decoys are available; hence, in that case, an alternative literature search is required for the comparative study.
- New vectors will be chosen to grow the fragment, and same analyses will be preceded.
Importance
In the drug discovery pipeline, chemoinformatics and structure-based drug design play a critical and crucial role. It not only aims to explore the molecular basis of activity of target protein but also to predict possible derivatives that would improve activity. The three main components could be 1) Filtering large libraries into a smaller set of predicted “active” compounds that can be readily tested experimentally. 2) Optimizing the hit to lead compounds. 3) Designing new compounds, either by “growing” fragment molecules. Thus, this research proposal aims to deliver the process to handle the “hit to lead” phase.
References
Andricopulo AD, Guido RV & Oliva G (2008) Virtual screening and its integration with modern drug design technologies. Current medicinal chemistry 15(1):37-46.
Bulfer SL, Jean-Francois FL & Arkin MR (2015) Making FBDD Work in Academia. Fragment-based Drug Discovery: Lessons and Outlook.
Chackalamannil S, Rotella D & Ward S (2017) Comprehensive Medicinal Chemistry III. Elsevier,
Cheng T, Li Q, Zhou Z, Wang Y & Bryant SH (2012) Structure-based virtual screening for drug discovery: a problem-centric review. The AAPS journal 14(1):133-141.
Cramer RD, Patterson DE, Clark RD, Soltanshahi F & Lawless MS (1998) Virtual compound libraries: a new approach to decision making in molecular discovery research. Journal of chemical information and computer sciences 38(6):1010-1023.
Erlanson DA (2011) Introduction to fragment-based drug discovery. Fragment-based drug discovery and X-ray crystallography, Springer. p 1-32.
Erlanson DA, Fesik SW, Hubbard RE, Jahnke W & Jhoti H (2016) Twenty years on: the impact of fragments on drug discovery. Nature Reviews Drug Discovery 15(9):605.
Fernández-de Gortari E, García-Jacas CR, Martinez-Mayorga K & Medina-Franco JL (2017) Database fingerprint (DFP): an approach to represent molecular databases. Journal of cheminformatics 9(1):9.
González-Medina M, Prieto-Martínez FD, Owen JR & Medina-Franco JL (2016) Consensus diversity plots: a global diversity analysis of chemical libraries. Journal of cheminformatics 8(1):63.
Hong H, Xie Q, Ge W, Qian F, Fang H, Shi L, Su Z, Perkins R & Tong W (2008) Mold2, molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics. Journal of chemical information and modeling 48(7):1337-1344.
Hu Y, Stumpfe D & Bajorath Jr (2011) Lessons learned from molecular scaffold analysis. Journal of chemical information and modeling 51(8):1742-1753.
Jhoti H (2008) Fragment-based drug discovery using rational design. Sparking Signals, Springer. p 169-185.
Johnson MA & Maggiora GM (1990) Concepts and applications of molecular similarity. Wiley,
Kaserer T, Beck KR, Akram M, Odermatt A & Schuster D (2015) Pharmacophore models and pharmacophore-based virtual screening: concepts and applications exemplified on hydroxysteroid dehydrogenases. Molecules 20(12):22799-22832.
Koutsoukas A, Paricharak S, Galloway WR, Spring DR, IJzerman AP, Glen RC, Marcus D & Bender A (2013) How diverse are diversity assessment methods? A comparative analysis and benchmarking of molecular descriptor space. Journal of chemical information and modeling 54(1):230-242.
Lavecchia A & Di Giovanni C (2013) Virtual screening strategies in drug discovery: a critical review. Current medicinal chemistry 20(23):2839-2860.
McGregor MJ & Muskal SM (1999) Pharmacophore fingerprinting. 1. Application to QSAR and focused library design. Journal of chemical information and computer sciences 39(3):569-574.
McGregor MJ & Muskal SM (2000) Pharmacophore fingerprinting. 2. Application to primary library design. Journal of chemical information and computer sciences 40(1):117-125.
O’Boyle NM & Sayle RA (2016) Comparing structural fingerprints using a literature-based similarity benchmark. Journal of cheminformatics 8(1):36.
Rogers D & Hahn M (2010) Extended-connectivity fingerprints. Journal of chemical information and modeling 50(5):742-754.
Shoichet BK (2004) Virtual screening of chemical libraries. Nature 432(7019):862.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Medicine"
The area of Medicine focuses on the healing of patients, including diagnosing and treating them, as well as the prevention of disease. Medicine is an essential science, looking to combat health issues and improve overall well-being.
Related Articles

DMCA / Removal Request
If you are the original writer of this research project and no longer wish to have your work published on the UKDiss.com website then please: