Quality of Recovery of Laparoscopic Bariatric Surgery Patients
Info: 9266 words (37 pages) Example Research Project
Published: 1st Dec 2021
Tagged: MedicineSurgical Studies
TITLE: A prospective study to describe the quality of recovery of morbidly obese patients following laparoscopic bariatric surgery.
Table of Contents
Click to expand Table of Contents
List of Abbreviations
PROTOCOL SUMMARY
1 Background and Rationale
1.1 Background
1.2 Rationale
2 Objectives and Hypotheses
2.1 Primary Objective(s) & Hypothesis(es)
2.2 Secondary Objective(s) & Hypothesis(es)
3 METHODOLOGY
3.1 Summary of Study Design
3.2 Study Population
3.3 Inclusion Criteria
3.4 Exclusion Criteria
3.5 Stratification
4 Endpoints and Dataset
4.1 Endpoints
4.2 Dataset
5 STUDY PROCEDURES
5.1 General Informed Consent
6 Safety Reporting and Related Procedures
6.1 Adverse Event Reporting
6.1.1 Investigator responsibility
6.2 DEFINITIONS
6.2.1 Adverse Event (AE)
6.2.2 Adverse Reaction (AR); also referred to as Adverse Drug Reaction (ADR)
6.2.3 Serious Adverse Event (SAE)/Serious Adverse Reaction (SAR)
6.2.4 Non-serious Adverse Reaction (NSAR)
6.2.5 Special Situations
6.2.6 Health Outcome of Interest (HOI)
6.2.7 Sponsor’s Product
6.2.8 Causality Assessment
6.3 Sponsor Responsibility for Reporting Adverse Events (Optional)
7 Statistical Analysis Plan
7.1 Statistical Methods
7.1.1 Primary Objective(s): Calculation of Epidemiological Measure(s) of Interest (e.g. descriptive statistics, hazard ratios, incidence rates, test/retest reliability)
7.1.2 Secondary Objective(s): Calculation of Epidemiological Measure(s) of Interest (e.g. hazard ratios, incidence rates, test/retest reliability)
7.2 Bias
7.2.1 Methods to Minimize Bias
7.2.2 Limitations
7.3 Sample Size and Power Calculations
8 ADMINISTRATIVE AND REGULATORY DETAILS
8.1 Confidentiality
8.1.1 Confidentiality of Data
8.1.2 Confidentiality of Subject Records
8.1.3 Confidentiality of Investigator Information
8.2 Compliance with Financial Disclosure Requirements
8.3 Compliance with Law, Audit and Debarment
8.4 Compliance with Study Registration and Results Posting Requirements
8.5 Quality Management System
8.6 Data Management
9 Publications
10 References
11 SIGNATURES
List of Abbreviations
| ADR | Adverse Drug Reaction |
| AE | Adverse Event |
| ASA | American Society of Anaesthesiologists |
| BD | Business Day |
| BMI | Body Mass Index |
| CD | Calendar Day |
| DSUR | Development Safety Update Report |
| EDC | Electronic Data Capture |
| EMEA | European Medicines Agency |
| GPP | Good Pharmacoepidemiology Practices |
| HDU | High Dependency Unit |
| HOI | Health Outcomes of Interest |
| ICD-10 | International Classification of Disease, 10th Modification |
| IEC | Independent Ethics Committee |
| IQR | Interquartile Range |
| IRB | Institutional Review Board |
| ITU | Intensive Care Unit |
| NHS | National Health Service |
| NMB | Neuromuscular Block |
| NSAR | Non-serious Adverse Reaction |
| PBRER | Periodic Benefit Risk Evaluation Report |
| PSUR | Periodic Safety Update Report |
| SAE | Serious Adverse Event |
| SD | Standard Deviation |
| SOP | Standard Operating Procedure |
PROTOCOL SUMMARY
1 Background and Rationale
1.1 Background
Morbidly obese patients undergoing laparoscopic bariatric surgery are particularly vulnerable to post-operative complications and require specifically tailored anaesthetic procedures1.
The administration of a neuromuscular blocking agent (NMBA) facilitates patient ventilation during surgery, prevents involuntary movements by the patient and enhances surgical exposure. Rucuronium, an aminosteroid and cisatacurium, a benzylisoquinone compound are two commonly used neuromuscular blocking agents with different molecular and pharmacological properties4. When surgery has been completed, reversal of NMBA activity allows patients to breathe independently and resume normal muscle function. Acetylcholine inhibitors like neostigmine or an amionsteroid encapsulating agent like sugammadex are used to rapidly reverse NMB agents. Rapid reversal of neuromuscular blockade after surgery is desirable to avoid postoperative residual curarization (PORC) which can lead to potentially fatal respiratory problems. Rapid and reliable reversal of NMB agents also improves pain management, patient comfort and recovery3,5,6.
Several studies have reported favourable outcomes with the use of sugammadex for the recovery of neuromuscular functions in morbidly obese patients7–9. In a randomised controlled trial comparing the effects of sugammadex and neostigmine in patients undergoing laparoscopic surgery, 94% (n=66) of patients treated with sugammadex recovered within 5 minutes compared with 20% (n=65) of patients treated with neostigmine8.
Despite encouraging evidence on the efficacy and safety of sugammadex, clinicians and healthcare resource managers require justification for the greater cost of this drug. One such justification may be the impact of this drug on the quality of post-surgical recovery of patients.
1.2 Rationale
Selection of appropriate NMB reversal agents plays an important role in managing successful post-surgical recovery of morbidly obese patients. With rising number of obese patients in the UK, surgical departments are increasingly under pressure to improve services, increase efficiency and also save money10. A study of the various anaesthetic procedures and the quality of recovery of morbidly obese patients following bariatric surgery will inform crucial decisions about surgical treatment pathways and help decrease post-surgical recovery times.
2 Objectives and Hypotheses
2.1 Primary Objective(s) & Hypothesis(es)
To describe the total time spent in PACU by morbidly obese patients following laparoscopic bariatric surgery.
2.2 Secondary Objective(s) & Hypothesis(es)
To describe:
- The recovery process of morbidly obese patients following laparoscopic bariatric surgery
- The anaesthetic procedures used in laparoscopic bariatric surgery for morbidly obese patients
- The demographic and clinical characteristics of patients undergoing laparoscopic bariatric surgery
3 METHODOLOGY
3.1 Summary of Study Design
This is a prospective observational study of morbidly obese patients who undergo laparoscopic bariatric surgery across the operating suites at three hospital sites in the UK over a period of 3 months. The choice of a prospective study model is to allow real-time documentation of surgical and anaesthetic procedures and post-operative recovery of patients at each hospital site.
Anonymized data will be collected both retrospectively from a review of the patient’s medical notes to determine the underlying diagnosis, medications taken and clinical characteristics, and prospectively from the point of administration of general anaesthesia prior to the operation to when they are transferred to the ward following surgery.
Data will be collected by nominated members of the NHS theatre team, recovery unit team, and ward staff at the point of care to minimize observer bias. Data will be collected directly onto paper data collection forms before being transcribed into an electronic database.
3.2 Study Population
The study population will include 120 morbidly obese patients undergoing laparoscopic bariatric surgery in three hospital sites in the UK over a period of three months.
3.3 Inclusion Criteria
- Morbidly obese patients (BMI 40kg/m2 or above) who are undergoing laparoscopic bariatric surgery
- Age 18 or over at the time of surgery
3.4 Exclusion Criteria
- Patients whose medical records are unavailable
- Patients that either refuse or lack capacity to provide written informed consent
- Patients with conditions that contraindicate the use of NMBA
3.5 Stratification
Stratification of data will be performed for the following variables:
- Type of NMB agent
- Type of NMB block (deep or intermediate)
- Type of NMB reversal agent
4 Endpoints and Dataset
4.1 Endpoints
Primary Endpoint:
Mean time in PACU according to type of NMBA, NMBA reversal agent and depth of neuromuscular block.
Secondary Endpoints:
Pathway to post-surgical recovery
- Mean duration of surgery (calculated from first incision to completion of last suture)
- Mean duration of anaesthesia (Time from pre-oxygenation of patient, tracheal intubation, induction/maintenance/reversal of anaesthesia up to tracheal extubation)
- Percentage of patients admitted to intensive therapy unit (ITU) and high dependency unit (HDU)
- Mean length of stay in ITU/HDU
- Mean overall length of stay in hospital
- Percentage of patients discharged earlier than planned
- Percentage of patients with pain and post-operative nausea and vomiting (PONV) (visual analog scale scores if available)
- Distribution of pain and PONV scores
- Distribution of train of four (TOF) ratios at time of admission to PACU
- Distribution of oxygen saturation levels for patients in PACU
- Percentage of patients with difficulty in swallowing after extubation
- Percentage of patients with evidence of residual neuromuscular block (based on neuromuscular monitoring and clinical assessment)
- Percentage of patients with evidence of PORC
- Mean time required by patients to get out of be independently post NMBA reversal
Anaesthetic procedures used in laparoscopic bariatric surgery
- Distribution of NMBA and NMBA reversal agents used
- Distribution of the reasons of choice for each NMBA/NMBA reversal agent
- Distribution of the level of neuromuscular block ( Deep/Intermediate)
- Percentage of patients with difficult intubations
- Distribution of choice of body weight type used to calculate anaesthetic dose ( Ideal body weight/Adjusted body weight)
Demographic and clinical characteristics of patients undergoing laparoscopic bariatric surgery
- Distribution of age, sex, body weight and BMI
- Description of medical history and current medications
- Description of comorbidities
- Distribution of ASA physical status scores before surgery
4.2 Dataset
The following data will be presented according to type of NMBA/NMBA reversal agent and stratified according to level of neuromuscular block (deep/moderate):
- Mean time in PACU according to type of NMBA, NMBA reversal agent and depth of neuromuscular block.
- Mean duration of surgery (calculated from first incision to completion of last suture)
- Mean duration of anaesthesia (Time from pre-oxygenation of patient, tracheal intubation, induction/maintenance/reversal of anaesthesia up to tracheal extubation)
- Percentage of patients admitted to intensive therapy unit (ITU) and high dependency unit (HDU)
- Mean length of stay in ITU/HDU
- Mean overall length of stay in hospital
- Percentage of patients discharged earlier than planned
- Percentage of patients with pain and post-operative nausea and vomiting (PONV) (visual analog scale scores if available)
- Distribution of pain and PONV scores
- Distribution of train of four (TOF) ratios at time of admission to PACU
- Distribution of oxygen saturation levels for patients in PACU
- Percentage of patients with difficulty in swallowing after extubation
- Percentage of patients with evidence of residual neuromuscular block (based on neuromuscular monitoring and clinical assessment)
- Percentage of patients with evidence of PORC
- Mean time required by patients to get out of be independently post NMBA reversal
- Distribution of NMBA and NMBA reversal agents used
- Distribution of the reasons of choice for each NMBA/NMBA reversal agent
- Distribution of the level of neuromuscular block ( Deep/Intermediate)
- Percentage of patients with difficult intubations
- Distribution of choice of body weight type used to calculate anaesthetic dose ( Ideal body weight/Adjusted body weight)
- Distribution of age, sex, body weight and BMI
- Description of medical history and current medications
- Description of comorbidities
- Distribution of ASA physical status scores before surgery
5 STUDY PROCEDURES
5.1 General Informed Consent
Written informed consent will be obtained from each patient before for enrolment in this study. No study data will be collected from any patients unless written consent has been provided.
Written informed consent will be sought by either members of the hospital research and development department or by members of the direct care team. Patients will be provided a copy of the information sheet detailing the study and its requirements. Adequate time will be granted to patients so that they have an opportunity to read the information sheet and to ask questions before any decision to take part is made and written consent sought.
A copy of the signed consent form will be maintained by the study investigator at each participating centre in the study site file.
6 Safety Reporting and Related Procedures
Introduction
This is a non-interventional study based on secondary use of data collected from healthcare professionals or consumers for other purposes. No administration of any therapeutic or prophylactic agent is required in this protocol, and there are no procedures required as part of this protocol.
6.1 Adverse Event Reporting
6.1.1 Investigator responsibility
Although adverse events are not actively solicited in this study, there are certain circumstances in which individual adverse events will be reported. For example, during review of medical records or physician notes (paper or electronic), to collect data as required by the protocol, if a notation of a serious adverse reaction (SAR), including death, or a non-serious adverse reaction (NSAR) to any MSD product is identified, the event must be reported according to Table 1.
Similarly, pre-specified Health Outcomes of Interest (HOIs) that meet criteria for SAR/NSAR, special situations, and any spontaneously reported AEs must be reported according to Table 1.
Table 1: AE Reporting Timeframes and Process for Investigators and Vendors
| EVENT TYPE | INVESTIGATOR TIMEFRAMES | VENDOR TIMEFRAMES |
| Investigator to Vendor [1], [2]
OR Investigator to MSD [3] |
Vendor to MSD
[4] |
|
| SAR
Pre-specified HOI that meets criteria of SAR Serious Special Situation, regardless of causality |
24 hours from receipt | 1 BD/3 CD from time of receipt from investigator |
| NSAR
Pre-specified HOI that meets criteria of NSAR Non-serious Special Situation, regardless of causality |
10 CD from receipt | 1 BD/3 CD from time of receipt from investigator |
| Spontaneously reported adverse events for MSD products-submit using above timeframes | ||
| If the investigator elects to submit AEs for non-MSD products, they should be reported to the market authorization holder (MAH) for that product or to the health authority according to the institution’s policy or local laws and regulations. | ||
| Follow-up to any event-submit using above timeframes | ||
| BD-Business Day; CD-Calendar Day | ||
| [1] AE reports from investigators must be transmitted via fax, secure email (if available), or entered directly into vendor’s electronic data collection (EDC) platform, if utilized.
[2] Investigator to Vendor: Applies to events for MSD products when a vendor is managing AE reporting from investigator to MSD. Events for MSD products are not entered in study database but must be forwarded to MSD for regulatory reporting. [3] Investigator to MSD: Applies to studies that do not have a vendor managing AEs. [4] Vendor to MSD: Applies to events for MSD products if the vendor is managing AE reporting between investigator and MSD. |
||
Submitting AE reports to MSD Global Safety: All AEs must be submitted via Fax to MSD UK Pharmacovigilance Department at 0032 2402 5990, in English using an AE form for reporting to worldwide regulatory agencies as appropriate.
6.2 DEFINITIONS
6.2.1 Adverse Event (AE)
Any untoward medical occurrence in a patient or clinical investigation subject administered sponsor’s product and which does not necessarily have to have a causal relationship with this product. An adverse event can therefore be any unfavourable and unintended sign (including an abnormal laboratory finding, for example), symptom, or disease temporally associated with the use of the product, whether or not considered related to the product. Any worsening (i.e., any clinically significant adverse change in frequency and/or intensity) of a preexisting condition that is temporally associated with the use of the product, is also an adverse event.
6.2.2 Adverse Reaction (AR); also referred to as Adverse Drug Reaction (ADR)
An AE which has a causal relationship with the product, that is, a causal relationship between the product and the adverse event is at least a reasonable possibility.
6.2.3 Serious Adverse Event (SAE)/Serious Adverse Reaction (SAR)
An adverse event or adverse reaction that results in death, is life threatening, results in persistent or significant disability/incapacity, requires inpatient hospitalization, prolongation of existing inpatient hospitalization, is a congenital anomaly/birth defect, or is another important medical event. Other important medical events that may not result in death, may not be life-threatening, or may not require hospitalization may be considered an SAE/SAR when, based upon appropriate medical judgment, they may jeopardize the patient or subject and may require medical or surgical intervention to prevent one of the other outcomes listed previously. Examples of such medical events include allergic bronchospasm requiring intensive treatment in an emergency room or at home and blood dyscrasias or convulsions that do not result in inpatient hospitalization.
6.2.4 Non-serious Adverse Reaction (NSAR)
An adverse reaction that does not meet any of the serious criteria in 6.2.3.
6.2.5 Special Situations
The following special situations are considered important safety information and must be reported, regardless of seriousness or causality, if the investigator becomes aware of them:
- Overdose
- Exposure to product during pregnancy or lactation
- Lack of therapeutic effect
- Off-label use, medication error, misuse, abuse, or occupational exposure
- Suspected transmission via a medicinal product of an infectious agent
6.2.6 Health Outcome of Interest (HOI)
Health Outcomes of Interest (HOIs) are pre-specified clinical events or outcomes that are collected according to the protocol. HOIs may be represented as diagnosis, treatment or procedures. Examples of HOIs include syncope or hypoglycaemia collected as study endpoints. HOIs must be assessed as part of AE collection and may meet criteria for AE reporting. Specifically, the investigator must assess each HOI for serious criteria and causality. If the HOI meets criteria specified in the protocol for AE reporting, then it must be reported as such.
6.2.7 Sponsor’s Product
Sponsor’s product includes any pharmaceutical product, biological product, device, diagnostic agent or protocol-specified procedure, whether investigational (including placebo or active comparator product) or marketed, manufactured by, licensed by, provided by or distributed by the Sponsor for human use.
6.2.8 Causality Assessment
A causality assessment is the determination of whether or not there is at least a reasonable possibility that a product caused the adverse event. Causality must be recorded on the AE form for each reported event in relationship to a Sponsor’s product.
Primary Data Collection
The assessment of causality is to be determined by an investigator who is a qualified healthcare professional according to his/her best clinical judgment. Use the following criteria as guidance (not all criteria must be present to be indicative of causality to a Sponsor’s product): There is evidence of exposure to the Sponsor’s product; the temporal sequence of the AE onset relative to the administration of the Sponsor’s product is reasonable; the AE is more likely explained by the Sponsor’s product than by another cause.
Secondary Data Collection
Only AEs with an explicit and definitive notation (by a healthcare provider) of a causal relationship with a product in the medical records or other secondary data being reviewed should be reported as NSAR/SARs. During review of secondary data, causality should never be assigned retrospectively.
6.3 Sponsor Responsibility for Reporting Adverse Events (Optional)
All adverse events will be reported to regulatory agencies, IRB/IECs and investigators in accordance with all applicable global laws and regulations.
7 Statistical Analysis Plan
7.1 Statistical Methods
All analyses will be descriptive in nature. Both distributions and descriptive statistics of both central tendency (medians and arithmetic or geometric means) and dispersion (standard deviation, interquartile range) will be presented for quantitative variables. Nominal variables will be described with frequencies and percentages while ordinal variables will also have medians and interquartile ranges described.
7.1.1 Primary Objective(s): Calculation of Epidemiological Measure(s) of Interest (e.g. descriptive statistics, hazard ratios, incidence rates, test/retest reliability)
Descriptive statistics and distribution of time in PACU will be presented for each NMB/NMB reversal agent according to depth of neuromuscular block.
7.1.2 Secondary Objective(s): Calculation of Epidemiological Measure(s) of Interest (e.g. hazard ratios, incidence rates, test/retest reliability)
Descriptive statistics and distributions will be presented for each secondary endpoint.
7.2 Bias
7.2.1 Methods to Minimize Bias
This is study is purely descriptive and aims to document specific anaesthetic procedures and associated quality of recovery of patients at a limited number of hospital sites in the UK. No comparative and/or generalizable conclusions are expected from this study.
7.2.2 Limitations
This study is limited to the description of neuromuscular block and reversal management practices and associated quality of recovery of 120 morbidly obese patients undergoing bariatric surgery in three hospital sites in the UK. As such, there is no randomization of patients as all morbidly obese patients that undergo the planned surgical procedure and requiring general anaesthesia will be included. This study will not generate generalizable knowledge.
7.3 Sample Size and Power Calculations
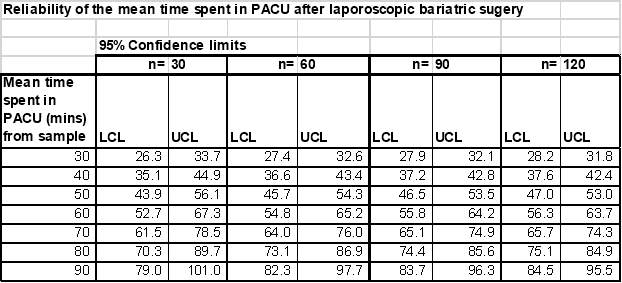
This evaluation will document data from 120 patients undergoing laparoscopic bariatric surgery at 3 hospital sites in the UK over a three month period. Number of hospital sites may be further expanded if the requisite number of patients is not available within the study period. The principal outcome of the study is to describe the mean time from admission to the Post Anaesthetic Care Unit (PACU) to discharge to a surgical ward. The table below shows the reliability of this outcome for various sample sizes and expected mean length of stay in PACU:
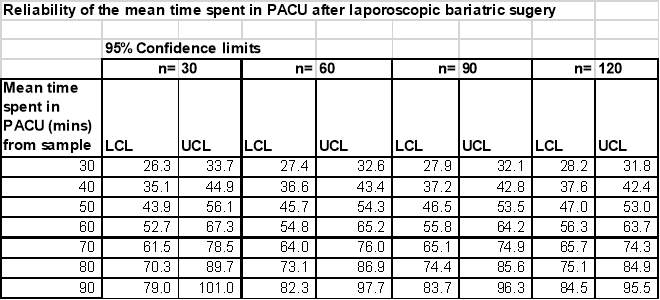
As patients will be stratified according to whether deep or moderate block was used, these are the numbers needed in each group.
Thus, a sample of 60 per group i.e. an overall sample of 120 , would provide sufficiently reliable outcomes for both each group separately and overall.
8 ADMINISTRATIVE AND REGULATORY DETAILS
8.1 Confidentiality
8.1.1 Confidentiality of Data
By signing this protocol, the investigator affirms to the Sponsor that information furnished to the investigator by the Sponsor will be maintained in confidence, and such information will be divulged to the Institutional Review Board, Ethics Review Committee or similar or expert committee; affiliated institution and employees, only under an appropriate understanding of confidentiality with such board or committee, affiliated institution and employees. Data generated by this study will be considered confidential by the investigator, except to the extent that it is included in a publication as provided in the Publications section of this protocol.
8.1.2 Confidentiality of Subject Records
By signing this protocol, the investigator agrees that the Sponsor (or Sponsor representative), Institutional Review Board/Independent Ethics Committee (IRB/IEC), or Regulatory Agency representatives may consult and/or copy study documents in order to verify worksheet/case report form data. By signing the consent form, the subject agrees to this process. If study documents will be photocopied during the process of verifying worksheet/case report form information, the subject will be identified by unique code only; full names/initials will be masked prior to transmission to the Sponsor.
By signing this protocol, the investigator agrees to treat all subject data used and disclosed in connection with this study in accordance with all applicable privacy laws, rules and regulations.
8.1.3 Confidentiality of Investigator Information
By signing this protocol, the investigator recognizes that certain personal identifying information with respect to the investigator, and all sub-investigators and study site personnel, may be used and disclosed for study management purposes, as part of a regulatory submissions, and as required by law. This information may include:
- name, address, telephone number and e-mail address;
- hospital or clinic address and telephone number;
- curriculum vitae or other summary of qualifications and credentials; and
- other professional documentation.
Consistent with the purposes described above, this information may be transmitted to the Sponsor, and subsidiaries, affiliates and agents of the Sponsor, in your country and other countries, including countries that do not have laws protecting such information. Additionally, the investigator’s name and business contact information may be included when reporting certain serious adverse events to regulatory agencies or to other investigators. By signing this protocol, the investigator expressly consents to these uses and disclosures.
If this is a multicenter study, in order to facilitate contact between investigators, the Sponsor may share an investigator’s name and contact information with other participating investigators upon request.
8.2 Compliance with Financial Disclosure Requirements
Financial Disclosure requirements are outlined in the US Food and Drug Administration Regulations, Financial Disclosure by Clinical Investigators (21 CFR Part 54). It is the Sponsor’s responsibility to determine, based on these regulations, whether a request for Financial Disclosure information is required. It is the investigator’s/subinvestigator’s responsibility to comply with any such request.
The investigator/subinvestigator(s) agree, if requested by the Sponsor in accordance with 21 CFR Part 54, to provide his/her financial interests in and/or arrangements with the Sponsor to allow for the submission of complete and accurate certification and disclosure statements. The investigator/subinvestigator(s) further agree to provide this information on a Certification/Disclosure Form, commonly known as a financial disclosure form, provided by the Sponsor or through a secure password-protected electronic portal provided by the Sponsor. The investigator/subinvestigator(s) also consent to the transmission of this information to the Sponsor in the United States for these purposes. This may involve the transmission of information to countries that do not have laws protecting personal data.
8.3 Compliance with Law, Audit and Debarment
By signing this protocol, the investigator agrees to conduct the study in an efficient and diligent manner and in conformance with this protocol; generally accepted standards of Good Pharmacoepidemiology Practice and all applicable federal, state and local laws, rules and regulations relating to the conduct of the study.
The investigator also agrees to allow monitoring, audits, Institutional Review Board/Independent Ethics Committee review and regulatory agency inspection of study-related documents and procedures and provide for direct access to all study-related source data and documents.
The investigator agrees not to seek reimbursement from subjects, their insurance providers or from government programs for procedures included as part of the study reimbursed to the investigator by the Sponsor.
The Investigator shall prepare and maintain complete and accurate study documentation in compliance with Good Pharmacoepidemiology Practice, standards and applicable federal, state and local laws, rules and regulations; and, for each subject participating in the study, provide all data, and, upon completion or termination of the study, submit any other reports to the Sponsor as required by this protocol or as otherwise required pursuant to any agreement with the Sponsor.
Study documentation will be promptly and fully disclosed to the Sponsor by the investigator upon request and also shall be made available at the investigator’s site upon request for inspection, copying, review and audit at reasonable times by representatives of the Sponsor or any regulatory agencies. The investigator agrees to promptly take any reasonable steps that are requested by the Sponsor as a result of an audit to cure deficiencies in the study documentation and worksheets/case report forms.
The investigator must maintain copies of all documentation and records relating to the conduct of the study in accordance with their institution’s records retention schedule which is compliant with all applicable regional and national laws and regulatory requirements. If an institution does not have a records retention schedule to manage its records long-term, the investigator must maintain all documentation and records relating to the conduct of the study for 5 years after final report or first publication of study results, whichever comes later, per GPP guidelines. This documentation includes, but is not limited to, the protocol, worksheets/case report forms, advertising for subject participation, adverse event reports, subject source data, correspondence with regulatory authorities and IRBs/ERCs, consent forms, investigator’s curricula vitae, monitor visit logs, laboratory reference ranges, laboratory certification or quality control procedures and laboratory director curriculum vitae. All study documents shall be made available if required by relevant regulatory authorities. The investigator must consult with the Sponsor prior to discarding study and/or subject files.
The investigator will promptly inform the Sponsor of any regulatory agency inspection conducted for this study.
Persons debarred from conducting or working on studies by any court or regulatory agency will not be allowed to conduct or work on this Sponsor’s studies. The investigator will immediately disclose in writing to the Sponsor if any person who is involved in conducting the study is debarred or if any proceeding for debarment is pending or, to the best of the investigator’s knowledge, threatened.
In the event the Sponsor prematurely terminates a particular study site, the Sponsor will promptly notify that site’s IRB/IEC.
According to European legislation, a Sponsor must designate an overall coordinating investigator for a multi-center study (including multinational). When more than one study site is open in an EU country, MSD, as the Sponsor, will designate, per country, a national principal coordinator (Protocol CI), responsible for coordinating the work of the principal investigators at the different sites in that Member State, according to national regulations. For a single-center study, the Protocol CI is the principal investigator. In addition, the Sponsor must designate a principal or coordinating investigator to review the study report that summarizes the study results and confirm that, to the best of his/her knowledge, the report accurately describes the conduct and results of the study in the study’s final report. The Sponsor may consider one or more factors in the selection of the individual to serve as the Protocol CI and or CSR CI (e.g., availability of the CI during the anticipated review process, thorough understanding of study methods, appropriate enrollment of subject cohort, timely achievement of study milestones). The Protocol CI must be a participating study investigator.
8.4 Compliance with Study Registration and Results Posting Requirements
By signing this protocol, the investigator agrees to conduct the study in an efficient and diligent manner and in conformance with this protocol; generally accepted standards of Good Pharmacoepidemiology Practice; and all applicable local laws, rules and regulations relating to the conduct of the clinical study.
The investigator also agrees to allow monitoring, audits, Institutional Review Board/Independent Ethics Committee review and regulatory agency inspection of study-related documents and procedures and provide for direct access to all study-related source data and documents.
The investigator agrees not to seek reimbursement from subjects, their insurance providers or from government programs for procedures included as part of the study reimbursed to the investigator by the Sponsor.
The Investigator shall prepare and maintain complete and accurate study documentation in compliance with Good Pharmacoepidemiology Practice, standards and applicable local laws, rules and regulations; and, for each subject participating in the study, provide all data, and, upon completion or termination of the study, submit any other reports to the Sponsor as required by this protocol or as otherwise required pursuant to any agreement with the Sponsor.
Study documentation will be promptly and fully disclosed to the Sponsor by the investigator upon request and also shall be made available at the investigator’s site upon request for inspection, copying, review and audit at reasonable times by representatives of the Sponsor or any regulatory agencies. The investigator agrees to promptly take any reasonable steps that are requested by the Sponsor as a result of an audit to cure deficiencies in the study documentation and worksheets/case report forms.
The investigator must maintain copies of all documentation and records relating to the conduct of the study in accordance with their institution’s records retention schedule which is compliant with all applicable regional and national laws and regulatory requirements. If an institution does not have a records retention schedule to manage its records long-term, the investigator must maintain all documentation and records relating to the conduct of the study for 5 years after final report or first publication of study results, whichever comes later, as per Good Pharmacoepidemiology Practice (GPP) guidelines. This documentation includes, but is not limited to, the protocol, worksheets/case report forms, advertising for subject participation, adverse event reports, subject source data, correspondence with regulatory authorities and IRBs/ERCs, consent forms, investigator’s curricula vitae, monitor visit logs, laboratory reference ranges, laboratory certification or quality control procedures and laboratory director curriculum vitae. All study documents shall be made available if required by relevant regulatory authorities. The investigator must consult with the Sponsor prior to discarding study and/or subject files.
The investigator will promptly inform the Sponsor of any regulatory agency inspection conducted for this study.
Persons debarred from conducting or working on studies by any court or regulatory agency will not be allowed to conduct or work on this Sponsor’s studies. The investigator will immediately disclose in writing to the Sponsor if any person who is involved in conducting the study is debarred or if any proceeding for debarment is pending or, to the best of the investigator’s knowledge, threatened.
In the event the Sponsor prematurely terminates a particular study site, the Sponsor will promptly notify that site’s IRB/IEC.
According to European legislation, a Sponsor must designate an overall coordinating investigator for a multi-center study (including multinational). When more than one study site is open in an EU country, MSD, as the Sponsor, will designate, per country, a national principal coordinator (Protocol CI), responsible for coordinating the work of the principal investigators at the different sites in that Member State, according to national regulations. For a single-center study, the Protocol CI is the principal investigator. In addition, the Sponsor must designate a principal or coordinating investigator to review the study report that summarizes the study results and confirm that, to the best of his/her knowledge, the report accurately describes the conduct and results of the study in the study’s final report. The Sponsor may consider one or more factors in the selection of the individual to serve as the Protocol CI and or CSR CI (e.g., availability of the CI during the anticipated review process, thorough understanding of study methods, appropriate enrollment of subject cohort, timely achievement of study milestones). The Protocol CI must be a participating study investigator.
8.5 Quality Management System
By signing this protocol, the Sponsor agrees to be responsible for implementing and maintaining a quality managementsystem with written development procedures and functional areastandard operating procedures (SOPs) to ensure that studies are conducted and data are generated, documented, and reported in compliance with the protocol, accepted standards of Good Pharmacoepidemiology Practice, and all applicable federal, state, and local laws, rules and regulations relating to the conduct of the study.
8.6 Data Management
Study data will be analysed using Microsoft Excel™ 2010 and Stata v14 (Stata Corp). Data received by [Vendor] will be checked for patient eligibility, accuracy and completeness using visual and programmed validation checks. Data queries will be raised directly with the participating Trust.
All data files will be securely stored on a password protected internal server at [Vendor] and all study-related documents will be kept in a locked cupboard when not in use. Access to the study database will be restricted (by password protection) and only granted to [Vendor] staff who are directly involved with the study.
The investigator or qualified designee is responsible for recording and verifying the accuracy of subject data. By signing this protocol, the investigator acknowledges that his/her electronic signature is the legally binding equivalent of a written signature. By entering his/her electronic signature, the investigator confirms that all recorded data have been verified as accurate.
For an outsourced study the institutional policies of the vendor should be followed for development of data management plans. However, the vendor should ensure compliance with Good Pharmacoepidemiology Practice, and all applicable federal, state, and local laws, rules and regulations relating to the conduct of the study
9 Publications
Study results will be disseminated as posters or oral presentations at appropriate clinical conferences, to be identified at the start of the study in collaboration with by clinical experts. The results will also be published in an end of study report for the Sponsor and submitted as a paper to a peer-reviewed academic journal.
10 References
1. Nightingale, C. E. et al. Peri-operative management of the obese surgical patient 2015. Anaesthesia 70, 859–876 (2015).
2. Ogunnaike, B. O., Jones, S. B., Jones, D. B., Provost, D. & Whitten, C. W. Anaesthetic considerations for bariatric surgery. Anesth. Analg. 95, 1793–1805 (2002).
3. Green, M. S., Venkatesh, A. G. & Venkataramani, R. Management of Residual Neuromuscular Blockade Recovery: Age-Old Problem with a New Solution. Case Rep. Anesthesiol. 2017, (2017).
4. Appiah-Ankam, J. & Hunter, J. M. Pharmacology of neuromuscular blocking drugs. (2004).
5. Hayes, A. H., Mirakhur, R. K., Breslin, D. S., Reid, J. E. & McCourt, K. C. Postoperative residual block after intermediate-acting neuromuscular blocking drugs. Anaesthesia 56, 312–318 (2001).
6. Brueckmann, B. et al. Effects of sugammadex on incidence of postoperative residual neuromuscular blockade: a randomized, controlled study. Br. J. Anaesth. 115, 743–751 (2015).
7. Abrishami, A., Ho, J., Wong, J., Yin, L. & Chung, F. Sugammadex, a selective reversal medication for preventing postoperative residual neuromuscular blockade. Cochrane Database Syst. Rev. (2009). doi:10.1002/14651858.CD007362.pub2
8. Geldner, G. et al. A randomised controlled trial comparing sugammadex and neostigmine at different depths of neuromuscular blockade in patients undergoing laparoscopic surgery*. Anaesthesia 67, 991–998 (2012).
9. Gaszynski, T., Szewczyk, T. & Gaszynski, W. Randomized comparison of sugammadex and neostigmine for reversal of rocuronium-induced muscle relaxation in morbidly obese undergoing general anaesthesia. Br. J. Anaesth. 108, 236–239 (2012).
10. Obesity: identification, assessment and management | Guidance and guidelines | NICE. Available at: https://www.nice.org.uk/guidance/cg189. (Accessed: 4th June 2017)
11 SIGNATURES
Sponsor’s Representative
TYPED NAME SIGNATURE DATE
_________________________ ____________________________ __________
Investigator
I agree to conduct this study in accordance with the design outlined in this protocol and to abide by all provisions of this protocol (including other manuals and documents referenced from this protocol); deviations from the protocol are acceptable only with a mutually agreed upon protocol amendment. I agree to conduct the study in accordance with generally accepted standards of Good Pharmacoepidemiology Practice. I also agree to report all information or data in accordance with the protocol and, in particular, I agree to report any serious adverse experiences as defined in Section 6 – Safety Reporting and Related Procedures. I understand that information that identifies me will be used and disclosed as described in the protocol, and that such information may be transferred to countries that do not have laws protecting such information. Since the information in this protocol is confidential, I understand that its disclosure to any third parties, other than those involved in approval, supervision, or conduct of the study is prohibited. I will ensure that the necessary precautions are taken to protect such information from loss, inadvertent disclosure, or access by third parties.
TYPED NAME SIGNATURE DATE
_________________________ ____________________________ __________
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Surgical Studies"
Surgery is the branch of medicine that involves the treatment of injuries, diseases and other conditions by operative methods, i.e. by cutting open the body and removing or repairing a damaged part. Technological advances mean that many surgeries can now be performed without large incisions using what is known as keyhole surgery.
Related Articles
DMCA / Removal Request
If you are the original writer of this research project and no longer wish to have your work published on the UKDiss.com website then please: