Towards the Preparation of an AAB Periodic Copolymer of Styrene and Maleimide Derivatives
Info: 15059 words (60 pages) Dissertation
Published: 11th Dec 2019
Tagged: Chemistry
Contents
1.1 Potential of sequence controlled polymers
1.2 Methods of sequence control
1.3 Alternating & periodic copolymers
Abstract
An ability to control the sequence of synthetic polymers holds incredible potential. This is apparent when looking at the almost unbelievable complexity achieved by sequence controlled polymers in nature, for example in DNA and proteins. It is therefore surprising that the field of sequence controlled polymers is currently understudied. The work carried out in this report aims to add to the relatively small number of sequence controlled polymers that have been produced through synthetic means by producing the first example of an AAB periodic copolymer of styrene and maleimide derivatives.
Recently, cyclopolymerisation has been introduced as a technique for preparing alternating and simple periodic copolymers. This uses controlled radical polymerisation methods, which provide unprecedented control over chain length and chain length distributions. In this project, a cyclisable monomer, containing two styrene units attached by hydrolysable bonds to a removable connecting unit, was designed and synthesised. This cyclisable monomer was cyclopolymerised with maleimide, taking advantage of the natural ability of styrene and maleimide to produce alternating copolymers. The concentration was optimised to ensure that the rate cyclisation was greater than intermolecular propagation. Analysis of the amount of unpolymerised alkene present in the polymer suggested that complete cyclopolymerisation appears to have taken place at the lowest concentrations. The removable connecting unit of the polymerised macrocycle was removed by hydrolysis.
Matrix assisted laser desorption/ionisation time-of-flight mass spectroscopy was used to analyse the polymer sequence. This confirmed the alternating structure prior to hydrolysis, but failed to obtain a spectrum of the hydrolysed polymers, presumably due to difficulty ionising the samples. This means conclusive evidence of a periodic copolymer was not obtained. Nevertheless, the desired periodic copolymer may have been produced. At the lowest concentrations complete cyclopolymerisation appeared to have taken place, and an alternating copolymer was produced. This was successfully hydrolysed which should give the AAB periodic copolymer. While this is encouraging, further work to obtain a spectrum must be carried out to be able to conclusively determine the sequence.
Abbreviations
1H NMR – Proton nuclear magnetic resonance
AFGP – Antifreeze glycoprotein
AIBN – azobisisobutyronitrile
APCI – Atmospheric pressure chemical ionization
ATRP – Atom transfer radical polymerisation
DCC – N,N-dicyclohexylcarbodiimide
DCM – Dichloromethane
DEMM-Br – diethyl 2-bromo-2-methyl malonate
DHU – dicyclohexylurea
DMAP – 4‑dimethylaminopyridine
ESI – Electrospray ionisation
GPC – Gel permeation chromatography
M1 – Monomer 1
M2 – Monomer 2
MALDI-TOF-MS – Matrix assisted laser desorption/ionisation time-of-flight mass
spectroscopy
Mw – Weight-average molecular weight
PDI – Polydispersity index
PLGA – Poly(lactic-co-glycolic acid)
PMI – N-propylmaleimide
RAI – Relative alkene integral
SET – Single electron transfer
THF – Tetrahydrofuran
TLC – Thin layer chromatography
1 Introduction
Polymer chemistry has become essential to modern life since the development of the first synthetic polymers over a century ago. Our ability to control polymer properties has come a long way; for example, relatively recent advancements such as the development of controlled polymerisation has provided us with unprecedented control of polymer chain length, composition and architecture.1 However, control of polymer microstructure (the precise ordering of repeating units within a copolymer chain) remains a challenging task in polymer science.
As is the case for many areas of chemistry, polymer chemists can look to nature for inspiration. Evolution over billions of years has created organisms of almost unimaginable complexity, and the remarkable ability of organisms to control polymer microstructures, such as in proteins and DNA, is essential to biological systems. With this in mind, it seems reasonable to say that controlling the microstructures of synthetic polymers surely holds incredible potential, and will be a huge milestone in polymer chemistry. Despite this, the field of sequence controlled polymers is currently understudied.
This report will highlight the importance of sequence controlled polymers and summarise the progress made so far, with a particular focus on alternating and simple periodic copolymers. The work carried out in this project aims to contribute to this fascinating area of research by synthesising a new AAB periodic copolymer, which will be explained in detail later in this report.
1.1 Potential of sequence controlled polymers
1.1.1 Control of bulk properties
The potential of controlling sequence in polymers extends further than simply the properties of materials, as will become apparent in this report; however, the connection between microstructure and bulk material properties should certainly not be overlooked. Polymer sequence has been found to have a direct impact on bulk properties, such as solubility2, heat resistance3,4, crystallisation5 and melting points6. Biodegradability of certain polymers can also be altered by changing the microstructure, and this has already found a role in useful real world applications. Poly(lactic-co-glycolic acid) (PLGA) is an important copolymer with various therapeutic applications, one of which is as a biodegradable drug-delivery encapsulation system. PLGA with an alternating copolymer sequence has been found to degrade in the body at a slower, more constant rate than the normally used random copolymer; this results in a smoother delivery of drugs to the patient which is desirable for drug-delivery systems.7
1.1.2 Polymer self-assembly and folding
The functions of proteins are essential to almost every biological process and arise from their precise 3-dimensional structure. Protein folding occurs due to the cumulative effect of relatively weak non-bonding interactions to give a minimum energy structure which is determined by the specific order of amino-acids.8 These folded structures have chemical capabilities far more advanced than what we can currently achieve with synthetic polymers, but there is certainly potential for polymers of the future to mimic protein capabilities.
The dominant driving force for protein folding and 3-dimentional structure determination is the hydrophobic effect. This effect describes the favourable process of excluding water from around hydrophobic non-polar amino acid residues, causing these non-polar residues to assemble in micelle-like fashion so they are in close proximity to other non-polar regions.9Other non-covalent interactions, such as hydrogen bonding and van der Waals interactions, play a lesser role in 3-dimentional structure determination10. Indeed,computational modelling has shown that protein folding can be almost entirely controlled by the ordering of polar and non-polar residues.11Further computational studies have built on this idea and shown that polymer folding can be determined simply by the sequence of polar and non-polar monomers in a copolymer chain.12
Research into the field of synthetic self-assembling macromolecules, known as “foldamers” has made progress in recent years.13 Some of these attempted foldamers have shown some self-assembling ability, but progress is mostly limited to short oligomers14, or repeating units which very closely resemble those used in nature, such as polypeptoids.5 As our ability to control polymer microstructure increases, the development of polymeric foldamers will inevitably gain momentum.
1.1.3 Coding macromolecules for information storage
DNA is a remarkable information storage system; all of the information required for life is coded into sequences consisting of four DNA bases. Just as DNA is a form of coded information storage, it is possible that a polymer sequence could be coded and subsequently interpreted to store and retrieve information. In a copolymer of two monomers “A” and “B”, each monomer can represent a “bit” of information, in the same way that computers use binary codes of 0 and 1.15
Very recently, examples of information storage and retrieval using short oligomers has been reported. Sequence controlled oligomers were produced and the microstructures were subsequently determined with tandem mass spectrometry and 1H NMR, providing proof of concept for information encoded into polymers and paving the way for more complex examples.16,17,18 This idea has been taken further by blending a sequence controlled oligomer into bulk industrially produced polymers (polystyrene and polymethacrylate); the oligomer sequence was decodable even when blended into these bulk polymers,opening up possibilities for anti-counterfeit methods or molecular barcodes.18,19
Despite these promising recent examples, synthesis and analysis of the primary sequence of copolymers is very difficult and is currently restricted to short oligomers.20Many challenges must be overcome before sequence controlled polymers become a viable method for information storage. The technology requires perfect control over polymer microstructure in much longer polymers, as well as chemical and analytic methods of reading and interpreting data in these polymer chains.
1.2 Methods of sequence control
1.2.1 Iterative addition
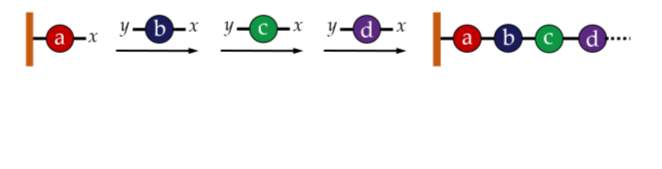 Perhaps the most obvious method for creating a sequence specific polymer is to add each desired monomer to the chain one-by-one using a standard chemical synthesis approach, as shown in figure 1. This method, known as iterative addition, was initially incredibly difficult and time consuming.21 The approach was dramatically improved by attaching the growing chain to a solid-phase support which allowed for much quicker and easier synthesis and purfication.22Over the years, developments have improved this technique further; for example, precise control of stereochemistry is now possible.23
Perhaps the most obvious method for creating a sequence specific polymer is to add each desired monomer to the chain one-by-one using a standard chemical synthesis approach, as shown in figure 1. This method, known as iterative addition, was initially incredibly difficult and time consuming.21 The approach was dramatically improved by attaching the growing chain to a solid-phase support which allowed for much quicker and easier synthesis and purfication.22Over the years, developments have improved this technique further; for example, precise control of stereochemistry is now possible.23
Unfortunately, despite considerable progress, this technique still delivers poor yield and efficiency, requires multiple extensive purification steps, and is not suitable for all monomers.24However, it remains a powerful tool for producing short sequence controlled oligomers.25
1.2.2 Step-growth polymerisation
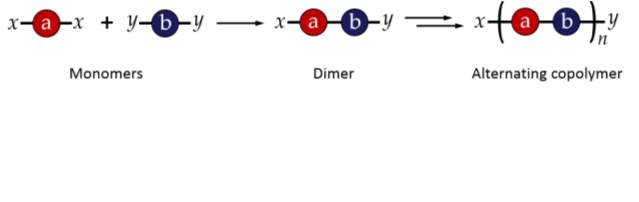 Step growth polymerisations involve monomers with two or more reactive functional groups. These functional groups react with functional groups on other monomers to give rise first to dimers, then short oligomers, and eventually long chain polymers. Step-growth polymerisation can be used to produce sequence controlled polymers by carefully selecting combinations of monomers which react with each other in a specific and predictable way.26 For example, if two monomers, one with two terminal “x” functional groups (x-a-x) and one with two “y” terminal functional groups (y-b‑y) are selected, where x preferentially reacts with y and vice versa, an alternating copolymer will be produced, as shown in figure 2.27
Step growth polymerisations involve monomers with two or more reactive functional groups. These functional groups react with functional groups on other monomers to give rise first to dimers, then short oligomers, and eventually long chain polymers. Step-growth polymerisation can be used to produce sequence controlled polymers by carefully selecting combinations of monomers which react with each other in a specific and predictable way.26 For example, if two monomers, one with two terminal “x” functional groups (x-a-x) and one with two “y” terminal functional groups (y-b‑y) are selected, where x preferentially reacts with y and vice versa, an alternating copolymer will be produced, as shown in figure 2.27
This can be extended to more complex copolymer structures by introducing different monomers sequentially; for example, an ABCDCBAE repeating sequence prepared with step-growth polymerisation has been reported, as shown in figure 3.28
Unfortunately, step-growth offers very little control over chain length and chain length distribution because polymer chains react to form new longer chains at random. This puts it at a disadvantage compared to some other polymerisation techniques.30
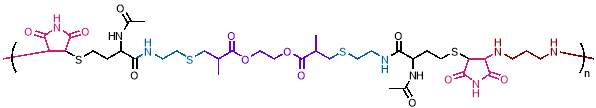
Figure 3 – Step growth polymerisation has been used to create complex periodic copolymers, as in this example with a ABCDCBAE repeating unit.
(Figure reproduced from reference 28)
1.2.3 Chain growth polymerisation
Chain growth polymerisation differs from step-growth polymerisation in that it works by continuously adding monomer units onto the end of a growing chain. Sequence control via chain growth polymerisation is inherently difficult, as it involves continuous and often indiscriminate reactions between highly reactive radical or anionic species which are difficult to control.31,32 Nevertheless, as will become apparent in this report, chain growth polymerisation has potential to be a powerful technique in sequence control.
Conventional “non-living” chain growth polymerisation suffers from a lack of control over chain length and chain length distribution due to the very high concentration of active chains which undergo frequent uncontrolled propagations and terminations.33For this reason, the relatively new development of controlled radical polymerisation (CRP) techniques, such as atom transfer radical polymerisation (ATRP)34, and single electron transfer (SET)35 are of particular interest to polymer chemists.
Controlled radical polymerisations offer unprecedented control over composition, chain length, chain length distribution and architecture.36The reasons for this are twofold. Firstly, the rate of initiation is much greater than rate of propagation, so much so that initiation is essentially instantaneous relative to propagation which allows all chains to begin growing at the same time.37 The second, and perhaps most important, reason is that termination of active chains is supressed. This is achieved by an equilibrium between dormant chains, which cannot propagate or terminate, and active propagating chains. The equilibrium massively favours dormant chains; therefore the concentration of active radical species at a given time is very low and termination of active chains is almost entirely eliminated.36 The result is that all chains grow at a very similar rate.
CRP provides an effective method for the preparation of copolymers with complex architectures, such as block and gradient copolymers, because polymer chains remain “alive” even after monomers have been used up.38 This means that when a new monomer is added to the reaction, the polymerisation can continue and the new monomer is added to the existing polymer chains.
Despite progress made in polymerisation techniques in recent years, CRP typically offers little control over polymer microstructure and copolymerisations usually give statistical copolymers.31Nevertheless, some monomer pairs naturally produce repeating microstructures and in recent years novel techniques employing CRP to produce simple regulated microstructures have had some success; this will be discussed later.39
1.3 Alternating & periodic copolymers
A periodic copolymer defines a microstructure with a regularly repeating pattern of two or more monomers. An alternating copolymer is the simplest periodic copolymer, where two monomer units repeat in alternating fashion i.e. (AB)n.
1.3.1 Importance of alternating and simple periodic copolymers
We cannot evaluate the properties of alternating or periodic copolymers if we cannot produce them, and due to our currently limited ability to produce such polymers we have only a very small number to investigate. In other words, we still have a lot to learn about the exact effect of an alternating or periodic polymer sequence on polymer properties.5 It is already known that small differences in microstructure can have quite large effects on bulk properties, and this has been utilised as long ago as the 1960’s when an alternating aromatic amide copolymer was found to have superior heat resistance compared to the equivalent random copolymer.3 It seems logical that many new alternating and periodic copolymers will also have useful enhanced properties compared to their random counterparts, so it is an important task to prepare such polymers and test their properties.
1.3.2 Applications as antifreeze agents
There is interesting potential for simple periodic copolymers to be used as antifreezing agents due to the importance of the AAB periodic structure in an interesting class of glycoproteins called antifreeze glycoproteins (AFGP). These glycoproteins consist of an AAB repeating structure of Alanine-Alanine-Threonine where the hydroxyl group of the threonine is modified with a disaccharide, as shown in figure 4.40 AFGPs are found in and northern cod and some Antarctic fish and form part of a wider class of antifreeze proteins which are vital to the survival of many organisms that live in sub‑zero conditions, from bacteria to plants and fish.41
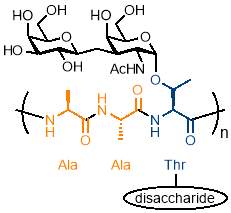
Figure 4 – Antifreeze glycoproteins consist of an AAB periodic copolymer of Alanine-Alanine-Threonine where the hydroxyl group of the threonine is modified with a disaccharide.
(Figure adapted from reference 41)
AFGPs work by binding to microscopic ice crystals to inhibit their further growth and therefore prevent macroscopic freezing. They are much more efficient than traditional anti-freezing agents such as ethylene glycol, and so required concentrations are orders of magnitude lower than their traditional counterparts. For this reason there is potential for AFGPs or synthetic derivatives to be used in many applications such as food-processing and cryopreservation.41
1.3.3 Preparing alternating and simple periodic copolymers

There are very few cases of monomer pairs with a natural tendency to form alternating copolymers using chain-growth methods, indeed the vast majority of CRP copolymerisations will give statistical copolymers.31Nevertheless, a few notable cases of highly selective naturally alternating copolymers do exist, for example with the case of styrene and maleimide which form a perfect alternating copolymer, as illustrated in figure 5.43
Figure 5 – Due to the large difference in electron density of the polymerisable bonds, styrene and maleimide will always form an alternating copolymer when polymerised.
Monomers pairs such as styrene and maleimide form alternating copolymers because of differences in the polarities of the polymerisable double bonds. A radical with high electron density on an electron rich styrene monomer will prefer to react with the electron deficient double bond of maleimide rather than another electron rich styrene double bond, and vice versa.42,44The result is an AB alternating copolymer. This process is so selective that even when one monomer is in vast excess, a near perfect alternating polymer will still be formed until one monomer is used up, at which point homopolymerisation of the remaining polymer will continue, albeit at a slower rate.45

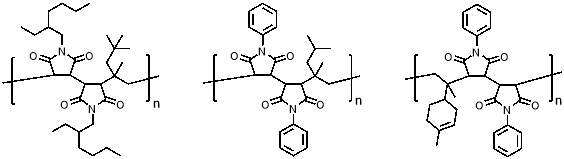 More examples of new alternating copolymers have recently been reported using increasingly complex monomers or conditions46,47. More excitingly, much rarer examples of simple periodic copolymers which have also been reported; for example, highly selective AAB periodic copolymers of N-substituted maleimides and different olefin monomers with high steric bulk (figure 6a and b).48 Similarly, the AAB periodic copolymer of N‑phenylmaleimide and d‑limonene when polymerised in a fluorinated alcohol solvent has been reported (figure 6c); this is thought to occur because of the combination of favourable interaction of carbonyls on adjacent maleimide units with the fluoroalcohol, and the bulkiness of the cyclic d-limonene structure.49
More examples of new alternating copolymers have recently been reported using increasingly complex monomers or conditions46,47. More excitingly, much rarer examples of simple periodic copolymers which have also been reported; for example, highly selective AAB periodic copolymers of N-substituted maleimides and different olefin monomers with high steric bulk (figure 6a and b).48 Similarly, the AAB periodic copolymer of N‑phenylmaleimide and d‑limonene when polymerised in a fluorinated alcohol solvent has been reported (figure 6c); this is thought to occur because of the combination of favourable interaction of carbonyls on adjacent maleimide units with the fluoroalcohol, and the bulkiness of the cyclic d-limonene structure.49
1.3.4 Cyclopolymerisation
Recently, controlled radical polymerisations involving a cyclopolymerisation step have been reported by the Sawamoto group as a promising method of preparing alternating and simple periodic copolymers. Cyclopolymerisation consists of two alternating propagation steps; an intramolecular cyclization followed by an intermolecular addition to form a polymer chain containing cyclic structures, as illustrated in figure 7.51
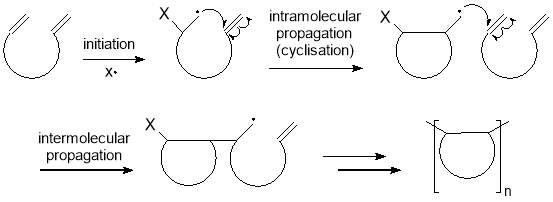
With cyclopolymerisation, two monomers that would typically form a random copolymer or separately homopolymerise can be encouraged to copolymerise in a predictable pattern. This is achieved by attaching the two monomers onto a “scaffold” so they are held in close proximity. Under CRP conditions, these monomers can undergo an intramolecular radical cyclisation at a rate that is much faster than an intermolecular propagation due to the close proximity of the two monomers. In order to obtain the desired polymer, the cyclic scaffold must be removed or opened via a cleavage process. Successful examples have been reported recently, which are summarised in figure 8 and discussed below.
Figure 7 – Cyclopolymerisation involves an intramolecular radical cyclisation followed by an intermolecular propagation to form a polymer chain containing cyclic structures.
One example (figure 8a) of cyclopolymerisation in sequence control used methacrylate (M) and acrylate (A) monomers which would typically form a random copolymer. The two monomers were attached to a naphthalene scaffold side by side at the 1,8 positions. A hydrolysable ester linkage was used to connect the monomers to the scaffold. Ruthenium catalysed CRP was used to promote cyclopolymerisation; this step was carried out very high dilution (1.0 mM) to ensure that the rate of radical cyclisation was much faster than intermolecular propagation to prevent unwanted cross-linking reactions. Methacrylate, being slightly more reactive than acrylate, reacts preferentially to the acrylate on the same monomer to give an alternating sequence. The naphthalene scaffold was removed by hydrolysis of the ester linkage to give an alternating copolymer in over 80% of the polymer chain, far greater than what would be seen in the statistical copolymer resulting from the regular polymerisation of these polymers.52 This is an impressive feat but still leaves room for improvement as the polymer chain consists of nearly 20% errors. The technique can be carried forward from this simple alternating example to more complex periodic copolymers.
Cyclopolymerisation was also used to create an alternating copolymer containing repeating carboxylic acid and alcohol pendant groups (figure 8b). First, a molecule containing a hemiacetal ester linkage between two vinyl moieties was synthesised. This molecule underwent ruthenium catalysed controlled radical cyclopolymerisation, and the hemiacetal ester linkage in the resultant cyclic structure was cleaved via hydrolysis to give an alternating copolymer with an error of less than 5%. This example is unique as it creates an alternating copolymer with two different pendant groups (i.e. carboxylic acid and alcohol).2
The first example of a periodic copolymer using cyclopolymerisation has also been reported (figure 8c), using a double cyclopolymerisation. Two styrene units were bonded to a tridentate ligand by hydrolysable amide bonds, to create a scaffold in a similar fashion to the previous examples. The structure was coordinated to palladium, and 4-vinyl pyridine was coordinated to the last vacant coordination site of the palladium metal centre. The result is three aromatic vinyl groups arranged side by side (styrene / vinyl pyridine / styrene). A double cyclopolymerisation under CRP conditions between three vinyl moieties gives what is effectively a bicyclic coordination structure. Removal of the palladium and hydrolysis of the amide linkages gave a well-defined ABA periodic copolymer with benzylamine (A) and pyridine (B) pendant groups.53 The novel use of cyclopolymerisation to produce a periodic copolymer can be investigated further and hopefully achieve the same result without the need for a heavy metal coordination centre.
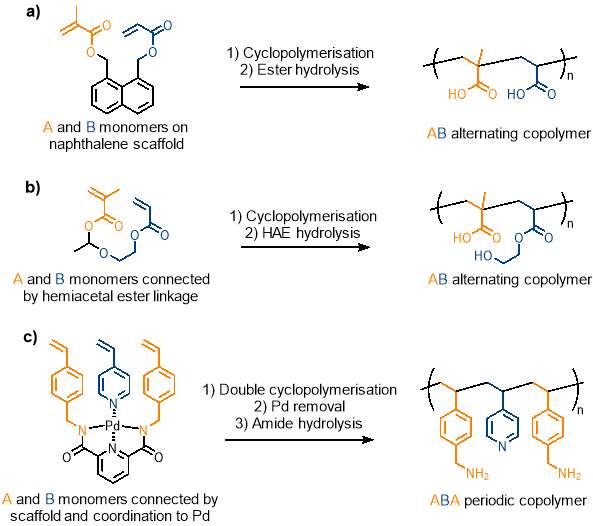
Figure 8 – Cyclopolymerisation has been used to produce alternating and periodic copolymers. a) cyclopolymerisation of acrylate monomers on a naphthalene scaffold; b) cyclopolymerisation of acrylate monomers connected by a hemiacetal ester linkage; c) double cyclopolymerisation of the ABA type monomer connected by a scaffold and by coordination to a Pd metal centre.
(Figure reproduced from reference 2)
1.4 This work
This work will develop the concept of cyclopolymerisation as a method of sequence control by working towards producing the first periodic copolymer using cyclopolymerisation without the need for a metal coordination centre. We aim to produce what will be, to our knowledge, the first example of an AAB periodic copolymer based on styrene and maleimide, as shown in 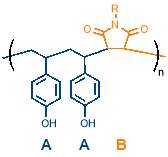
figure 9, adding to the small number of periodic copolymers that have so far been reported in literature.
1.5
Figure 9 – This work aims to produce the first example of an AAB periodic copolymer based on styrene and maleimide.

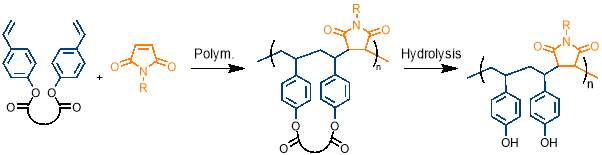
Our strategy combines cyclopolymerisation with the natural ability of styrene and maleimide to form highly selective alternating sequences. First, a monomer capable of undergoing cyclopolymerisation will be synthesised; this will contain two styrene units attached by cleavable bonds to a connecting unit. This monomer will be polymerised with maleimide, taking advantage of the alternating nature of maleimide with the styrene-derived monomer to give an alternating copolymer containing a macrocycle. Removal of the connecting unit will afford the desired alternating copolymer. This process is outlined in figure 10.
2 Results and discussion
2.1 Monomer design
Essential to the outcome of this project was the design of a suitable styrene derived monomer capable of cyclopolymerisation. The monomer must contain two styrene units, orientated in space in such a way as to allow a cyclization to occur; the monomer must also be rigid enough to hold the styrene moieties in this position. This is achievable by connecting the styrene units to a “connecting unit”. In order to obtain the final periodic copolymer, this connecting section must be removable. Taking inspiration from the monomer of the previously mentioned example of cyclopolymerisation by the Sawamoto group52, we opted for the same hydrolysable ester group.
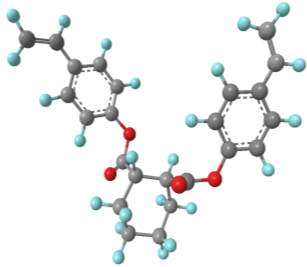
Computational modelling software (Chem3D Pro) was used to assess the three dimensional shape of the connecting units, and specifically how the two polymerisable bonds of the styrene were orientated relative to each other. Several plausible designs were considered, and eventually two, shown in 11, were selected to take forward.
Three-dimensional analysis of the monomers show that the polymerisable bonds meet the main requirement of being held relatively close to one another to ensure cyclisation is possible. Both monomers consist of two styrene units connected via an ester linkage to the 1,2‑ positions of a cyclic connecting unit; these connecting units are cyclohexane in the case of monomer 1 (M1) and benzene in monomer 2 (M2). Cyclic connecting units were selected to prevent rotation of the connecting bonds, therefore holding the styrene units in the required position.
| Monomer | Structure | 3D Structure |
| “M1” |  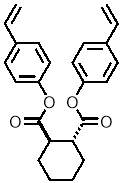 |
 |
| “M2” | 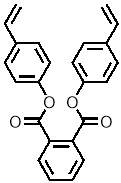 |
 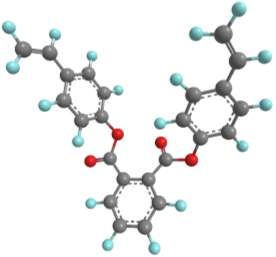 |
The most significant difference between the two monomers is that the cyclohexane of M1 will have greater flexibility than the aromatic ring of M2; this may benefit cyclization as it allows the molecule to accommodate the slight contortion necessary to achieve the exact orientation required for the reaction.
Figure 11 – Two monomers, M1 and M2 were selected for this project. Both structures hold the polymerisable bonds in close proximity to favour cyclisation, which is illustrated by the blue arrow.
During the monomer design process, it was important to ensure the ring size of the resulting cyclopolymerisation was as small as practically possible to favour the cyclisation step. With this in mind, the final monomer design has the styrene units connected directly to the ester, which are themselves connected side by side directly to the connecting unit, resulting in a 17‑member ring after cyclisation.
2.2 Monomer synthesis
The synthetic method required to produce each monomer was considered during the design process, therefore the two selected monomers have straightforward synthetic routes from relatively cheap and readily available starting materials. This section will discuss the procedure used in the synthesis of each monomer, including detailed mechanisms.
2.2.1 Synthesis of M1
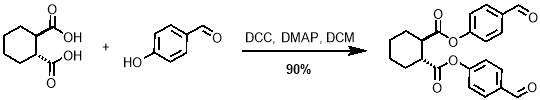
2.2.1.1 Steglichesterification
The first step in the synthesis of M1 is known as a Steglich esterification; it is a standard procedure for the formation of an ester from an alcohol and a carboxylic acid.54 The acid in this case was trans-1,2-cyclohexandicarboxylic. Ideally, the alcohol would be 4‑vinylphenol, however the high electron density on the alkene of this molecule, due to electron donation through the aromatic ring from the oxygen atom, make it highly reactive and therefore unsuitable for this reaction. For this reason, 4‑hydroxybenzaldehyde was selected over the more convenient 4‑vinylphenol, making a second step necessary to convert the aldehydes to alkenes. The acid and alcohol were reacted with N,N-dicyclohexylcarbodiimide (DCC) and 4‑dimethylaminopyridine (DMAP) which acts as a catalyst, as outlined in scheme 1. The reaction gave yields of up to 90%.
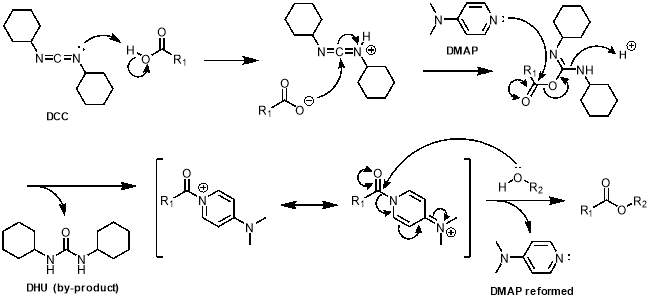
The mechanism of this reaction is shown in scheme 2. The process begins with deprotonation of the carboxylic acid by the nitrogen lone pair of DCC (step 1). This activates the imine of DCC and allows the newly formed carboxylate to attack at the imine carbon, forming an ester intermediate (step 2). The nitrogen in the aromatic ring of DMAP attacks the activated ester, kicking out the stable dicyclohexylurea (DHU) by-product, which precipitates out of the reaction (step 3). This gives a resonance stabilised species with a carbonyl attached to DMAP, allowing the alcohol to attack the carbonyl to form the desired ester (step 4), with DMAP being a good leaving group.
Scheme 1 – The first step in the synthesis of M1 is the Steglich esterification

2.2.1.2 Wittig reaction
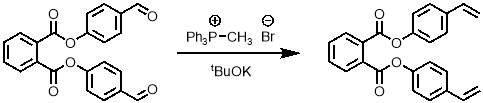
To obtain the desired monomer, the aldehydes of the bis-aldehyde intermediate must be converted into alkenes. The Wittig reaction was the obvious first choice for this transformation. A standard method for a Wittig reaction on another aromatic aldehyde from literature55 was used as shown in scheme 3.
Scheme 3 – The Wittig reaction was used to convert the bis-aldehyde into the bis-alkene.
Unfortunately, the conversion of the aldehydes to the alkenes was not as straightforward as expected. Initially, the reaction was carried out in standard (i.e. non-dry) dichloromethane (DCM). In these reactions, the conversion of aldehyde to alkene, which was monitored by 1H NMR, took place only very slightly (~2%) after several days. Instead, the 1H NMR showed the presence of an unexpected peak at a slightly lower chemical shift than the aldehyde of the intermediate, as shown in figure 12. The chemical shift (9.81 ppm) suggests this peak also corresponds to an aldehyde. It was theorised that this new aldehyde peak could be a result of hydrolysis of the ester of the bis‑aldehyde intermediate, which would form 4‑hydroxybenzaldehyde. To confirm this, thin layer chromatography (TLC) of the reaction mixture was run alongside a sample of 4‑hydroxybenzaldehyde; the TLC spots of the reaction indicated 4‑hydroxybenzaldehyde was present. Additionally, the 1H NMR obtained from the reaction mixture was compared with a sample of 4‑hydroxybenzaldehyde, this confirmed that 4‑hydroxybenzaldehyde was responsible for the new aldehyde peak.
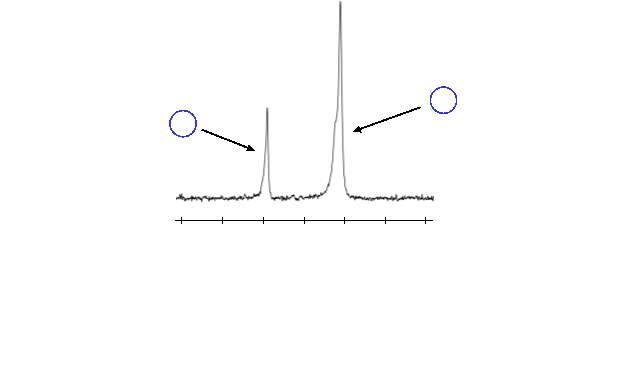
 The hydrolysis of the ester was likely due to the reaction of the strong base (tBuO‒) with water in the solvent, producing hydroxide ions that hydrolysed the ester by way of a nucleophilic attack. To overcome this problem, the reaction was repeated under anhydrous conditions; dry DCM was used in baked glassware, under a dry nitrogen atmosphere. This solved the ester hydrolysis problem; however, conversion of the aldehyde was still unacceptably low at ~6% after 48 hours.
The hydrolysis of the ester was likely due to the reaction of the strong base (tBuO‒) with water in the solvent, producing hydroxide ions that hydrolysed the ester by way of a nucleophilic attack. To overcome this problem, the reaction was repeated under anhydrous conditions; dry DCM was used in baked glassware, under a dry nitrogen atmosphere. This solved the ester hydrolysis problem; however, conversion of the aldehyde was still unacceptably low at ~6% after 48 hours.
δ (ppm)
The solvent (DCM) was swapped for dry tetrahydrofuran (THF), another common solvent used in the Wittig reaction. The reaction was repeated on a small scale achieving a reasonable yield of 68%, with 1H NMR of the reaction showing ~86% aldehyde to alkene conversion. With this promising result, the reaction was scaled up approximately 6-fold; however this larger scale reaction failed to achieve any conversion after 24 hours and was stopped. Initially it was thought that perhaps the problem lay in the difference in scale of the reaction, however several subsequent small-scale reactions were carried out using the same method, and these also failed to achieve any significant conversion. Due to time constraints of the project, the decision was made to set aside this synthesis and focus on synthesis of M2.
The cause of the problem is unknown, but it could be attributed to the age of the reactants used. Even more confusing is why one reaction achieved 86% conversion, while none of the others achieved more than 10%. If time allowed, an alternative method for the synthesis of M1 would have been attempted, involving the conversion of the carboxylic acid groups to acyl chloride by SOCl2, followed by a simple nucleophilic addition by 4-vinylphenol on this acyl chloride.
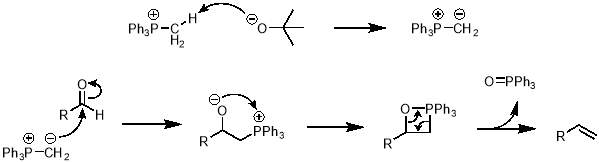
The mechanism of the Wittig reaction is shown in scheme 4. First, the tBuO‒ deprotonates the methyl of the methyltriphenylphosphonium ion to give an ylide. The newly formed carbanion can attack the electrophilic carbon of the aldehyde to give an intermediate with an adjacent positive and negative charge. This intermediate can undergo a ring closing reaction to neutralise both charges, leaving a strained 4-member ring. This ring can rearrange to give the desired alkene and triphenyl phosphine oxide as a stable by-product.
Scheme 4 – In this project the Wittig reaction was used to convert an aldehyde into an alkene, via the mechanism shown here.
2.2.2 Synthesis of M2
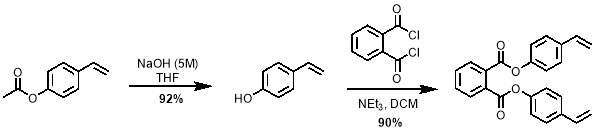
The synthesis of M2 features a simple nucleophilic attack of 4-vinyl phenol on a 1,2‑phthaloyl dichloride. However, 4-vinyl phenol is difficult to purchase and store due to its high reactivity. Therefore, the compound was produced from the hydrolysis of 4‑acetoxystyrene by NaOH shortly before use. It was then reacted with 1,2‑phthaloyl dichloride to give M2 in very good yields of up to 90%; the reaction is shown in scheme 5.
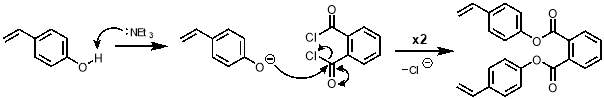
The mechanism for the synthesis of M2 from 4-vinylphenol is shown in scheme 6. The first step is the deprotonation of the phenol making it an excellent nucleophile. A nucleophilic attack by this anion on each acyl chloride of phthaloyl dichloride takes place to give M2.
Scheme 5 – 4-vinylphenol was prepared from the hydrolysis of 4-acetoxystyrene. This was then reacted with phthaloyl dichloride to give M2.
Scheme 6 – The synthesis of M2 proceeds viaa nucleophilic addition of deprotonated 4‑vinylphenol on each of the acyl chlorides of phthaloyl dichloride.
2.3 Polymerisation
This project will focus on a controlled radical method of polymerisation due to the high degree of control these techniques offer of polymer architecture and chain length. Selecting a suitable polymerisation technique and initiator, and optimising the polymerisation conditions was therefore an important part of this project. N-propylmaleimide was arbitrarily selected as the maleimide derived monomer.
2.3.1 Selecting an initiator
Selection of an initiator is important in any controlled radical polymerisation as it can influence chain length dispersity, rate of polymerisation and polymer architecture.56 In this project however, the initiator choice has added significance in the analysis of the polymer sequence.
Insection 2.5 of this report,the process of polymer sequence analysis is discussed in detail. As a brief introduction, it involves fragmentation of the polymer chain and analysis of the fragments with mass spectrometry. In a perfectly alternating or periodic copolymer, a repeating series of equally spaced peaks will be observed, with the difference between each subsequent peak corresponding to the molecular weight of one monomer on the end of the polymer chain. The complexity of the spectra can be significantly reduced if the first monomer in the chain (i.e. attached to the initiator) is controlled.

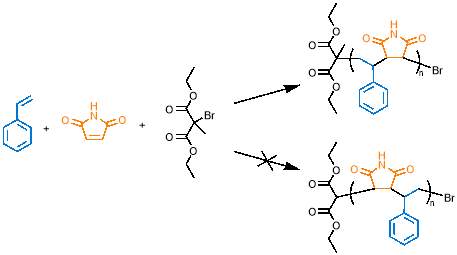
Following their work on sequence controlled polymers, the Sawamoto group has recently reported an alkyl bromide initiator, diethyl 2-bromo-2-methyl malonate (DEMM-Br), which favours initiation of styrene over maleimide with high selectivity57, as shown in figure 13. For this reason, DEMM-Br was selected as the initiator for polymerisations in this project.
The observed selectivity arises because the radical forms on an electron deficient carbon of the initiator, due to the two adjacent electron-withdrawing groups; this radical favours a reaction with the electron rich styrene monomer rather than the electron deficient maleimide.57
2.3.2 Trial polymerisations
2.3.2.1 Single electron transfer CRP
Selecting a suitable polymerisation technique and optimising the polymerisation conditions was an important part of this project. Initially, a controlled radical polymerisation technique first reported in 2006, known as single electron transfer (SET) CRP, was considered. This choice was made because SET-CRP has many advantages over other CRP techniques, but has not yet been extensively reported in literature. A considerable advantage of SET-CRP is that it is less sensitive to oxygen than many other CRP techniques; for this reason, it does not require a freeze-pump-thaw process to remove oxygen. Additionally, it employs relatively mild conditions and can be used at room temperature while still producing incredibly high molecular weight polymers very quickly.35
The mechanism behind this copper mediated polymerisation is centred around the instantaneous disproportion of Cu(I) into active Cu(0) and dormant Cu(II). The Cu(0) species initiates the dormant polymer chains by an outer sphere single electron transfer, while the Cu(II) deactivates the propagating chains, as is necessary in a controlled polymerisation.58 This is illustrated in figure 14.
Preliminary polymerisations found that although styrene could be easily polymerised following a literature method59, styrene and N‑propyl maleimide failed to co-polymerise under similar conditions. The polymerisation of styrene and maleimide by SET-CRP has not previously been reported in literature, and several further attempts using different conditions were unsuccessful. The decision was made to use an alternative polymerisation method.
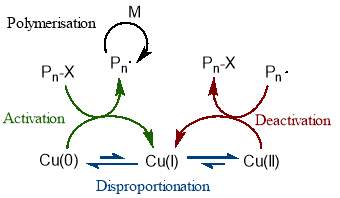
Figure 14 – In SET-CRP, Cu(I) instantaneously disproportionates into Cu(I) which activates dormant chains, and Cu(II) which deactivates propagating chains.
2.3.2.2 Atom-transfer radical polymerisation
Atom-transfer radical polymerisation is a robust and extensively studied CRP technique, making it a good choice for this project following the unsuccessful SET-CRP polymerisations. ATRP was first reported in 1995, and has since become one of the most successful and widely used CRP techniques.1
The mechanism of ATRP can be simplified to an equilibrium between active polymer chains and dormant chains mediated by a transition metal complex, as shown in figure 15. Many redox active metal centres can be used, but copper coordinated to nitrogen containing ligands is by far the most common.60 In the radical generating step (using a copper complex as an example), the Cu(I) metal centre undergoes a one electron oxidation to Cu(II) and simultaneously abstracts a halogen from a dormant polymer chain (or alkyl halide initiator) in what is known as an inner sphere electron transfer.61 Propagation steps between the radical species and monomer units can now occur, until the transfer of the halide from a Cu(II) complex back to the active radical on the polymer chain reforms the Cu(I) complex and leaves a dormant 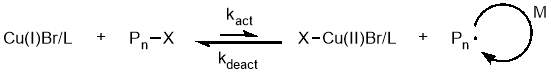
polymer chain.
Figure 15 – The mechanism of ATRP can be simplified to an equilibrium between active propagating chains and dormant species.
A polymerisation of styrene with N-propylmaleimide was successfully carried out using conditions adapted from literature43 (molar ratios of M2/PMI/CuBr/I/L[*] = 100/100/1/1/1.1, at 12 mol dm-3 in anisole), achieving 48% conversion in 4.5 hours. This promising result meant that the ATRP method could be taken forward and optimised for use in this project.
2.3.3 Cyclopolymerisation
The next stage in this project was the cyclopolymerisation of monomer M2 with N‑propylmaleimide to give an alternating copolymer.
2.3.3.1 Concentration
The most important consideration when selecting polymerisation conditions was monomer concentration, as this directly affects the degree of cyclopolymerisation and controls cross‑linking between polymer chains. Cyclopolymerisation has two key reaction rates to consider: the rate of intramolecular cyclisation and the rate of intermolecular propagation. For a successful cyclopolymerisation to take place, the rate of cyclisation must be much greater than the rate of intermolecular propagation. This ensures that cyclisation occurs before the intermolecular propagation step, giving rise to the macrocycle containing copolymer (figure 16, left). If the rate of intermolecular propagation is similar to or greater than cyclisation, the cyclisable monomer will form a pendant group because only one alkene is polymerised into the copolymer chain (figure 16, right); the pendant group would contain an unpolymerised alkene that can react further, as highlighted by the red circle in figure 16, causing cross-linking. The faster the cyclisation relative to intermolecular propagation, the less pendant group  formation will be seen.
formation will be seen.
The rates can be controlled by monomer concentration; cyclisation is a first order reaction, as it depends only on the concentration of cyclisable monomer M2, whereas the intermolecular step depends on the concentration of both the cyclisable monomer M2 (or cyclised monomer) and N‑propylmaleimide PMI, meaning this is a second order reaction. The rate equations are as follows:
Rate of cyclisation= kintraM2
Rate of intermolecular propagation = kinterM2PMI
Due to the difference in order, the rate of intermolecular propagation decreases much faster than cyclisation when monomer concentration is reduced. Therefore, to optimise cyclopolymerisation, the concentration of the polymerisations should be ideally as low as possible. However, if the concentration is too low the overall polymerisation will not take place, or will proceed very slowly. It was therefore necessary to find a concentration that provides an acceptable rate of polymerisation while maximising cyclopolymerisation.
2.3.3.2 Measurement of degree of cyclopolymerisation
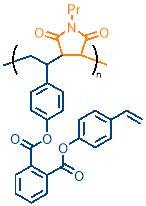
 As stated previously and illustrated in figure 16, a polymer where cyclopolymerisation has taken place will not contain any alkenes. Conversely, if complete cyclopolymerisation has not taken place, pendant groups containing an alkene will be present. These alkenes give a characteristic peak in the 1H NMR of the polymer, providing an easy method to assess the extent of cyclopolymerisation. The integral of a large unchanging section of the 1H NMR, corresponding to aromatic protons, was taken as a reference. The integral of the alkene peak was compared to the aromatic integral to give a relative alkene integration (RAI) value. This is illustrated in figure 17, which uses the polymer marked as polym. 4 in table 1 as an example.
As stated previously and illustrated in figure 16, a polymer where cyclopolymerisation has taken place will not contain any alkenes. Conversely, if complete cyclopolymerisation has not taken place, pendant groups containing an alkene will be present. These alkenes give a characteristic peak in the 1H NMR of the polymer, providing an easy method to assess the extent of cyclopolymerisation. The integral of a large unchanging section of the 1H NMR, corresponding to aromatic protons, was taken as a reference. The integral of the alkene peak was compared to the aromatic integral to give a relative alkene integration (RAI) value. This is illustrated in figure 17, which uses the polymer marked as polym. 4 in table 1 as an example.
This method cannot provide an absolute value for the extent of cyclopolymerisation as the broad 1H NMR peaks of the polymers are difficult to quantify, but rather it gives a value of cyclopolymerisation relative to the other polymers. Additionally, errors could arise if cross‑linking reactions of the pendant alkenes takes place, as this would reduce the integral of the alkene peak. If cross-linking does occur, it should be evident by the increased viscosity and lack of solubility of the resultant polymer.
2.3.3.3 ATRP Cyclopolymerisation attempts
| Polym. | [Monomer]
/ mol dm-3 |
Temp / °C | Time | Conversion | RAI | Mw | PDI |
| 1 | 1.00 | 90 | 7 days | 8% | – | – | – |
| 2 | 1.33 | 90 | 7 days | 18% | – | – | – |
| 3 | 2.00 | 90 | 7 days | 40% | – | – | – |
| 4 | 1.33 | 100 | 20 hours | 15% | 0.14 | 5804 | 1.67 |
| 5 | 0.75 | 100 | 90 hours | 25% | 0.10 | 5433 | 1.64 |
| 6 | 0.50 | 100 | 90 hours | 21% | 0.09 | 5874 | 1.66 |
| 7 | 0.25 | 100 | 90 hours | 15% | 0.08 | 6097 | 1.81 |
Final adjustments to the molar ratio of reactants were made in an attempt to decrease the resulting chain length for the purposes of easier analysis with mass spectrometry. The final molar ratio was M2/PMI/CuBr/I/L = 25/25/1/1.1/1.2 in anisole. For each polymer, gel permeation chromatography (GPC) was used to calculate weight-average molecular weight (Mw), and polydispersity index (PDI). PDI is a measure of the distribution of polymer chain lengths; a value of 1 represents a perfectly monodisperse polymer, while numbers greater than 1 represent increasing levels of polydispersity. The results of cyclopolymerisations 1‒7 are presented in table 1 and discussed below.
Table 1 – ATRP of M1 and PMI was carried out with a molar ratio of M1/PMI/CuBr/I/L = 25/25/1/1.1/1.2 at various concentrations. Monomer conversion, relative alkene integral (RAI), average molecular weight and PDI were calculated.
Initially, three polymerisations were carried at 1.00 M, 1.33 M and 2.00 M (polym. 1, 2 and 3 respectively) at 90 °C. These polymerisations progressed very slowly, achieving between 8% and 40% conversion after 7 days, at which point the polymers were precipitated. The 1H NMR of the polymerisations appeared to show the polymer peaks corresponding to N‑propylmaleimide are much greater than those of M2. This suggests that N‑propylmaleimide is polymerising much faster than M2, possibly because the activation energy of polymerisation for M2 is too high for polymerisation of this monomer to occur at a reasonable rate.
To overcome this problem, another polymerisation was carried out at 1.33 M (polym. 4) with the temperature increased from 90 °C to 100°C to provide the reaction with more energy to overcome the activation energy barrier. The polymerisation proceeded much faster, reaching 15% conversion after 24h, compared to just 8% in the 90°C polymerisation at the same concentration. Additionally, the 1H NMR showed a greater extent of incorporation of M2 into the polymer compared to the 90 °C sample. The 1H NMR also features significant alkene polymer peaks, which were found to have a relative alkene integration of 0.14. This value can be compared with relative alkene integration values for other polymers synthesised in this project, but should ideally be as close to zero as possible.
The presence of the alkene suggests cyclopolymerisation is not fully taking place, and therefore three polymerisations at lower concentrations of 0.75, 0.5 and 0.25 M (polym. 5, 6 and 7) were carried out, achieving between 15% and 25% conversion after 90 hours. The 1H NMR of all three polymers still showed significant alkene peaks, with RAI values of 0.10, 0.09 and 0.08 for polymerisations 5, 6 and 7 respectively. Of the four polymerisations carried out at 100 °C, a downward trend can be seen for the amount of alkene present, providing evidence for the favourable effect of lower concentration on cyclopolymerisation.
The molecular weight of these polymers is low with an average chain length of 10-12 repeat units. The PDI is high for a typical controlled radical polymerisation; however, this does not cause any problems for the outcome of the project.
The presence of the alkene in these polymers is problematic because if the concentration is lowered much further, the rate of polymerisation will become unreasonably slow. For this reason, it was decided than an uncontrolled method of polymerisation that would provide faster rates of polymerisation at low concentrations should be tested.
2.3.3.4 AIBN initiated cyclopolymerisation attempts
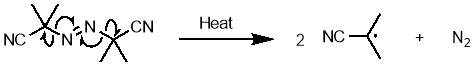
An uncontrolled polymerisation initiated by azobisisobutyronitrile (AIBN) was selected as an appropriate polymerisation method, due to its simplicity and known compatibility with maleimide and styrene.62 AIBN undergoes a thermal decomposition at temperatures above ~65 °C; homolytic cleavage of the weak C-N bond gives two carbon centred radicals that can go on to react with monomers to begin polymerisation, along with nitrogen gas. The structure and initiation mechanism of AIBN is shown in scheme 7.
Scheme 7 – AIBN undergoes thermal decomposition to give two carbon centred radicals and nitrogen gas.
Two polymerisations were conducted with a reactant molar ratio of M2/PMI/AIBN = 50/50/1 in anisole, at concentrations of 0.10 and 0.05 M. This reactant ratio keeps the ratio of initiator to monomer the same as previous ATRP reactions, as each AIBN molecule gives two radical species. The results of these polymerisations are presented in table 2.
| Polym. | [Monomer]
/ mol dm-3 |
Temp / °C | Time | Conversion | RAI | Mw | PDI |
| 8 | 0.10 | 75 | 4 hours | 70% | 0.01 | 467009 | 5.77 |
| 9 | 0.05 | 75 | 4 hours | 60% | 0.00 | 93649 | 2.28 |
As anticipated, the rate of polymerisation was much faster for these AIBN initiated polymerisations than ATRP despite using lower concentrations, reaching 70% and 60% conversion for 0.10 M and 0.05 M respectively in just 4 hours. Promisingly, the sample showed virtually no alkene peak, suggesting efficient cyclopolymerisation had occurred. However, decreased solubility compared to previous polymers was observed which could indicate that some cross-linking may have taken place, potentially giving a misleadingly low RAI value.
Table 2 – AIBN initiated radical polymerisation of M1 and PMI was carried with a molar ratio of M1/PMI/AIBN = 50/50/1 at two concentrations. Monomer conversion, relative alkene integral (RAI), average weight and PDI were calculated.
The PDI and molecular weight of these polymerisations were considerably higher than the previous controlled polymerisations. This is due to the uncontrolled nature of the polymerisations, which allows polymer chains of any length to terminate with any others, producing a large distribution of chain lengths, some of which can be extremely large. It was noted during the sequence analysis process that the mass spectra obtained from the uncontrolled polymerisations were nosier than the others, possibly due to the high PDI and molecular weight.
2.4 Hydrolysis
In order to obtain the desired AAB repeating unit, the connecting section of the cyclised M2 section of the polymer had to be removed via hydrolysis of the ester groups. Hydrolysis of polymers 4 ‒ 9 using lithium hydroxide was carried out. 1H NMR was used to assess whether removal of the connecting section had taken place. The appearance of sharp peaks after hydrolysis corresponding to aromatic protons of free connecting unit were observed, indicating they were no longer part of the polymer chains. The removal of the connecting unit appeared 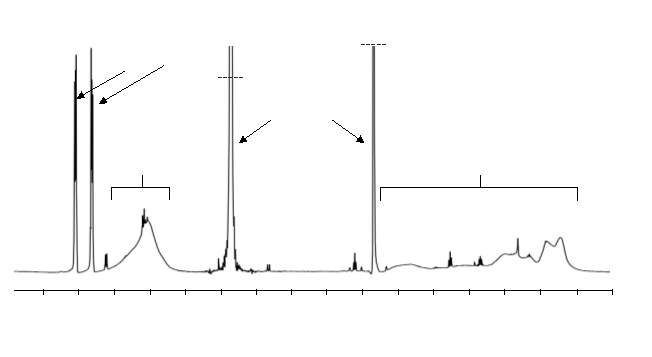 to be successful for all hydrolysed polymers.
to be successful for all hydrolysed polymers.
Figure 18 – 1H NMR was used to confirm hydrolysis had taken place; the presence of sharp aromatic proton peaks, separate from the broad polymer peaks, indicate successful hydrolysis.
2.5 Sequence analysis
Mass spectrometry was used to analyse the sequence of each polymer before and after hydrolysis. In each spectrum, a series of regularly spaced peaks should be observed, with the spacing between subsequent peaks in a series corresponding to the molecular weight of one monomer. This can be used to determine the ordering of monomers within the polymer chain.
Initially, matrix assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI‑TOF-MS) was used, as it is a very soft ionisation technique with high sensitivity and resolution, and is widely used for polymer sequence analysis.63
2.5.1 Pre-hydrolysis
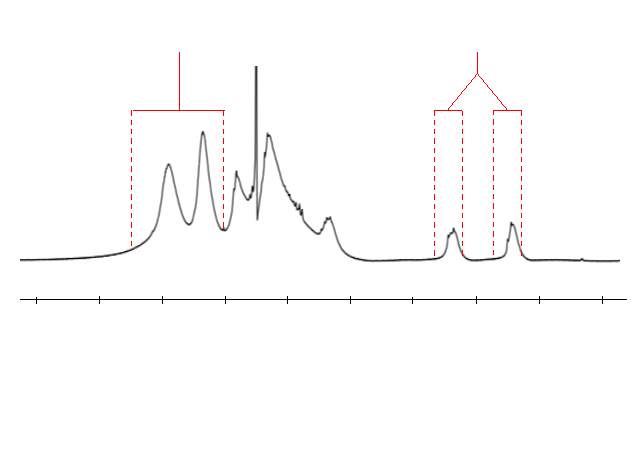
Prior to hydrolysis, polymers 4 ‒ 9 were analysed to confirm the alternating copolymer sequence. The spectrum of polymer 4 is shown as an example in figure 19, and the process of analysis is described below.
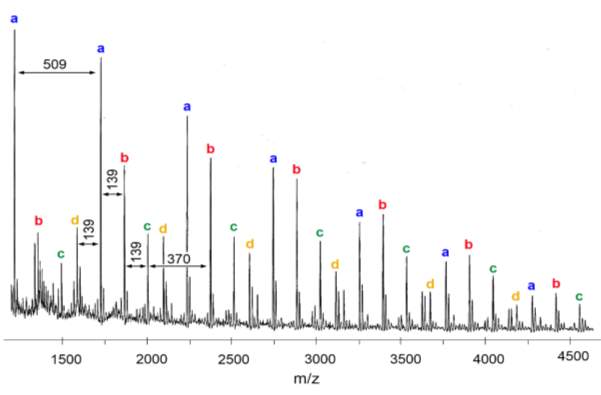
Figure 19 – MALDI-TOF-MS was used to analyse the sequence of polymers 4 ‒ 9. This figure shows the spectrum of polymer 4.
In figure 19, several series of peaks, each containing 4 peaks (labelled a‒d), have been identified. Each peak in a series has an m/z difference of 509 from the same peak in the subsequent series, which represents the mass of the repeat unit of PMI + M2. This is convincing evidence for the existence of an alternating copolymer sequence. The presence of multiple of peaks in a series (i.e. peaks a‒d) represent minor differences between the polymer chains. Each peak can be seen as having an m/z difference of 139 or 370 from another peak, corresponding to PMI (molecular weight 139.2) or M2 (molecular weight 370.4). Therefore, each peak in a series is the polymer chain differing by one or two monomers. The number of each monomer in the first four peaks of the spectrum of figure 19 has been calculated from the m/z value and this is shown in table 3. In the spectrum, the m/z value for each peak includes the polymer chain, the initiator and a Na+ adduct; the bromine has been eliminated by the laser, as seen in literature examples of MALDI polymer analysis.57
| Peak | m/z | M2 | PMI |
| a* | 1213 | 2 | 2 |
| b* | 1352 | 2 | 3 |
| c* | 1491 | 2 | 4 |
| d* | 1583 | 3 | 2 |
Table 3 – MALDI-TOF-MS was used to analyse the sequence of polymers 4 ‒ 9, the m/z value was used to calculate the number of each monomer present for each of the first four peaks, as shown here.
For a* and d*, the polymer presumably starts with M2 due to the styrene selective initiator used, and features an alternating copolymer chain ending with PMI for a* or M2 for d*, as would be expected. For peaks b* and c*, the number of units of PMI is greater than expected; this could be due to PMI starting the chain and ending the chain, which would mean the styrene selective initiator has not worked as expected, or it could be that two units of PMI have homopolymerised at some point in the polymer chain. Regardless, the incremental increase of 509 m/z between each peak in a series confirms an alternating structure for the majority of the polymer. The spectra for all analysed polymers showed a similar pattern to figure 19, suggesting all polymers have formed an alternating copolymer. This data cannot tell us whether the cyclopolymerisation has taken place; in order to determine this, the hydrolysed polymers must be analysed.
2.5.2 Post-hydrolysis
After hydrolysis, if the AAB periodic copolymer had successfully been synthesised, a repeating series of peaks similar to pre-hydrolysis should be observed. The m/z difference between equivalent peaks in each series would be ~381 rather than 509 as before, corresponding to the mass of the AAB repeat unit.
Mass spectrometry analysis of the hydrolysed polymers proved to be a very challenging task. Analysis by MALDI-TOF-MS was attempted several times, but failed to provide a spectrum with any significant peaks. Mass spectrometry using two other soft ionisation techniques, electrospray ionisation (ESI) and atmospheric pressure chemical ionisation (APCI), were also attempted several times, however these were also unsuccessful at obtaining a usable spectrum. It is unclear where the problem lies, although it appears that no detectable ions are being formed in the mass spectrometer. This is surprising as spectra prior to hydrolysis were easily obtained, and the presence of hydroxyl groups on the hydrolysed polymers should make it capable of ionisation, if anything more so than the pre-hydrolysed polymers. Regardless of the reason, no information about the sequence of the hydrolysed polymers was obtained from mass spectrometry.
3 Conclusion
A cyclisable monomer (M2) consisting of two styrene units attached by hydrolysable bonds to a connecting unit was designed using 3D computational modelling, and successfully synthesised. Polymerisations of this monomer with N-propylmaleimide afforded an alternating copolymer, as proven by sequence analysis with MALDI-TOF-MS. A second monomer (M1) was also designed but synthesis was unsuccessful and after failed several attempts attention was focused elsewhere.
Polymerisations were carried out at a range of concentrations, and the extent of cyclopolymerisation in each case was assessed by measuring the relative amount of alkene present in the polymer. As expected, cyclisation was maximised by lowering the concentration of the polymerisation to favour intramolecular cyclisation over intermolecular propagation. At the lowest concentrations, an AIBN initiated polymerisation was used because the rate of ATRP became impractically slow.
Hydrolysis of the polymers to remove the connecting section of the cyclisable monomer appeared to be successful. In the 1H NMR spectrum, new signals corresponding to the removed aromatic connecting unit were identified, while the broad peaks of the rest of the polymer remained.
Unfortunately, mass spectrometry failed to provide a spectrum for the hydrolysed polymers despite many attempts with different ionisation techniques. This means conclusive evidence for the AAB periodic copolymer was not obtained. Despite this, other evidence does suggest the desired periodic copolymer may have been produced. In the polymerisation at the lowest concentration, no alkene was observed in the polymer, which, assuming cross-linking did not occur to a significant extent, indicates that cyclopolymerisation was successful. This polymer showed an alternating sequence and hydrolysis appeared to have successfully removed the connecting unit. If these statements are correct, the resulting polymer would be the AAB periodic copolymer. While this is certainly encouraging, the sequence must be confirmed by mass spectrometry before a definite conclusion can be made.
3.1 Future work
short term..
Try with cyclohexane –
Extend to more complex periodic copolymers.
Replace styrene / maleimide with other monomers?
4 Experimental
4.1 General
All reagents and solvents were purchased from Aldrich Chemicals, with the following exceptions: PMDTA (Merck Millipore), trans-1,2-cyclohexandicarboxylic acid (Alfa Aesar), tetrahydrofuran (Fisher Scientific) and dichloromethane (Fischer Scientific). All reagents were used without further purification, with the exception of styrene, which was run over basic alumina prior to use to remove the polymerisation inhibitor.
Thin layer chromatography was performed on Merck precoated silica gel plates (0.25 mm, 60 F254) and viewed under UV light. Column chromatography was carried out using Aldrich silica gel with 60 Å pore size and 40-63 µm particle size.
4.2 Measurements
Molecular weights of polymers were measured in THF at 35 °C by gel permeation chorography on a Viscotek GPCmax VE 2001 chromatograph (flow rate 1.0 mL min-1) coupled to a Viscotek VE 3580 refractive index detector. The system was calibrated against a polystyrene standard.
1H NMR (400 MHz and 500 MHz) and 13C NMR (101 MHz) spectra were recorded on a Bruker Ascend spectrometer (equipped with a cryoprobe) using TMS as an internal reference.
Infrared spectra were measured on a Thermo Scientific Nicolet iS5 FT-IR Spectrometer.
MALDI-TOF-MS was performed on a Shimadzu Axima Confidence spectrometer equipped with a 337 nm nitrogen laser and used a dithranol matrix. ESI-MS was performed on a Walters SQ Detector 2. APCI was performed on a Thermo Scientific Exactive Plus EMR.
4.3 Monomer Synthesis
4.3.1 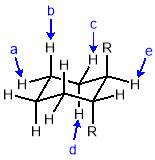
Synthesis of M1‑intermediate
A solution of 4-hydroxybenzaldehde (3.05 g, 25.0 mmol, 2.5 eqv), trans-1,2-cyclohexandicarboxylic acid (1.72 g, 25 mmol, 1 eqv.), N,N′-Dicyclohexylcarbodiimide (5.16 g, 25 mmol, 2.5 eqv.) and 4-dimethylaminopyridine (0.122 g, 1 mmol, 0.1 eqv.) in dichloromethane (50 mL) was stirred under nitrogen for 20 hours at room temperature. The mixture was then filtered to remove a solid white by-product. The resulting filtrate was washed with HCl (15 mL, 1 M) and saturated NaHCO3 (15 mL) before being dried over anhydrous MgSO4. The solvent was removed under reduced pressure to give a crystalline solid which was purified by column chromatography (SiO2, petroleum ether/ethyl acetate 3:2) to give M1‑intermediate (3.41 g, 90%) as an off-white crystalline solid. 1H NMR (400 MHz, CDCl3) δ: 10.01 (s, 2H, Hh), 7.93 (d, J = 8.5 Hz, 4H, Hg), 7.27 (d, J = 10.3 Hz, 4H, Hf), 3.01 (m, 2H, He), 2.41 (m, 2H, Hd), 1.99 (m, 2H, Hc), 1.64 (m, 2H, Hb), 1.49 (m, 2H, Ha); 13C NMR (101 MHz, CDCl3) δ: 190.84, 134.54, 133.54, 131.76, 130.57, 130.43, 128.70, 128.59, 122.29, 67.98, 44.95, 31.24, 25.62; IR (vmax/cm-1): 2932, 2860, 2361, 1747, 1690; MS (ESI+/‒): m/z = 403.2 [M+Na+].
4.3.2 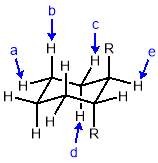
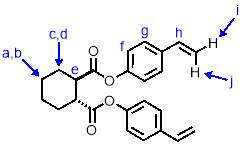
Synthesis of M1
In baked glassware, a solution of potassium tert-butoxide (590 mg, 10.52 mmol, 4 eqv.) and methyltriphenylphosphonium bromide (1.41 g, 7.89 mmol, 3 eqv.) in dry THF (5 mL) was cooled in an ice bath under nitrogen. To this cooled solution was added dropwise with stirring a solution of M1-intermediate (500 mg, 1.31 mmol, 1 eqv.) in dry THF (4 mL) which was prepared in baked glassware under nitrogen. After addition, the reaction was allowed to warm to room temperature and stirred for 24h. The reaction mixture was filtered to remove the solid precipitate by-product. The solvent was removed under reduced pressure and the resulting solid was dissolved in dichloromethane. This solution was washed with water (3 x 5 mL), and the aqueous phases extracted with dichloromethane. The organic phases were combined and dried over MgSO4, then evaporated under reduced pressure to give a viscous yellow oil. This was purified by column chromatography (SiO2, petroleum ether/ethyl acetate 9:1) to give M1 (340 mg, 68%) as a viscous, pale yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.31 (d, J = 8.6 Hz, 2H, Hg), 6.94 (d, J = 8.6 Hz, 2H, Hf), 6.62 (dd, J = 17.6, 10.9 Hz, 1H, Hh), 5.62 (d, J = 17.6 Hz, 2H, Hi), 5.16 (d, J = 10.9 Hz, 2H, Hj), 2.89 (m, 2H, He), 2.27 (m, 2H, Hd), 1.86 (m, 2H, Hc), 1.52 (m, 2H, Hb), 1.36 (m, 2H, Ha); 13C NMR (101 MHz, CDCl3) δ: 173.45, 150.23, 135.38, 127.17, 121.58, 121.55, 114.00, 45.00, 28.86, 28.05, 25.19; IR (vmax/cm-1): 3098, 2933, 2858, 2321, 1749, 1695 ; MS (ESI+): m/z = 399.2 [M+Na+].
4.3.3 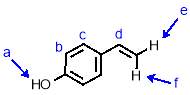
Synthesis of 4-vinyl phenol
To a stirred solution of 4-acetoxystyrene (2.19 g, 13.5 mmol, 1 eqv.) in THF (17 mL) was added with stirring NaOH solution (13.5 mL, 5 M). The reaction was stirred for 2 hours before being quenched by neutralisation with HCl (5 M). The product was extracted with ethyl acetate (3 x 35 mL) and the organic fractions combined and dried over MgSO4. The solvent was removed under reduced pressure to give 4-vinyl phenol (1.88 g, 92%) as a white crystalline solid. 1H NMR (400 MHz, CDCl3) δ: 7.23 (d, J = 8.5 Hz, 2H, Hc), 6.72 (d, J = 8.4 Hz, 2H, Hb), 6.58 (dd, J = 17.6, 10.9 Hz, 1H, Hd), 5.53 (d, J = 17.6 Hz, 1H, Hf), 5.05 (d, J = 10.9 Hz, 1H, He), 4.91 (b, 1H, Ha); 13C NMR (101 MHz, CDCl3) δ: 155.31, 136.14, 130.63, 127.60, 115.38, 111.63; IR (vmax/cm-1): 3379 (br), 3087, 2999, 1897, 1811, 1704, 1605, 1509; MS (ESI+): m/z = 145.0 [M+Na+].
4.3.4 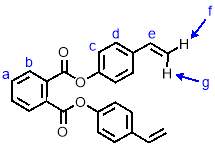
Synthesis of M2
To a solution of 4-vinyl phenol (1.62 g, 13.5 mmol, 2.5 eqv.) and triethylamine (1.64 g, 16.2 mmol, 3 eqv.) in dichloromethane (55 mL) was added dropwise with stirring 1,2-phthaloyl dichloride (1.10 g, 5.4 mmol, 1 eqv.). The reaction was left to run at room temperature for 16 hours, at which point it was washed with saturated sodium hydrogen carbonate solution (3 x 40 mL) and brine (40 mL). The solution was then dried over MgSO4 and the solvent removed under reduced pressure to give a pale yellow crystalline solid. This was purified by column chromatography (SiO2, petroleum ether/ethyl acetate 6:1) to give M2 (1.80 g, 90%) as an off-white crystalline solid. 1H NMR (400 MHz, CDCl3) δ: 7.99 (dd, J = 5.7, 3.3 Hz, 2H, Hb), 7.70 (dd, J = 5.7, 3.3 Hz, 2H, Ha), 7.43 (d, J = 8.6 Hz, 2H, Hd), 7.19 (d, J = 8.6 Hz, 4H, Hc), 6.71 (dd, J = 17.6, 10.9 Hz, 2H, He), 5.72 (d, J = 17.6 Hz, 2H, Hf), 5.26 (d, J = 10.9 Hz, 2H, Hg); 13C NMR (101 MHz, CDCl3) δ: 165.83, 150.25, 135.88, 135.68, 131.79, 131.67, 129.56, 127.29, 121.61, 114.20, 30.95; IR (vmax/cm-1): 2361, 2281, 1732, 1504; MS (ESI+): m/z = 393.2 [M+Na+].
4.4 Polymerisation
4.4.1 SET-CRP Polymerisation
A typical procedure for a SET-CRP polymerisation of styrene and N-propyl maleimide was as follows. A 25 mm length of copper wire was wrapped around a magnetic stirrer bar and washed in concentrated HCl and then acetone, then set aside under nitrogen. To a baked glass tube was added styrene (0.45 g, 4.37 mmol, 230 eqv.), N-propyl maleimide (0.61 g, 4.37 mmol, 230 eqv.), diethyl 2-bromo-2-methylmalonate (4.6 mg, 0.019 mmol, 1 eqv.), toluene (0.5 mL), PMDTA (1.1 mg, 0.0065 mmol, 0.35 eqv.) and copper (II) bromide (0.4 mg, 0.0018 mmol, 0.1 eqv.). The stirred bar and copper wire was added and the tube sealed with a septum cap. The solution was degassed by bubbling nitrogen through for 30 minutes. The reaction was stirred at 90 °C for several hours and monitored with 1H NMR.
4.4.2 ATRP
A typical procedure for an ATRP reaction of N-propyl maleimide and M2 was as follows. To a baked glass tube was added copper (I) bromide (3 mg, 0.021 mmol, 1 eqv.) and a magnetic stirrer bar. The tube was sealed with a septum cap and placed under nitrogen by three vacuum/nitrogen cycles. In a separate baked glass tube was added N-propyl maleimide (69.5 mg, 0.50 mmol, 25 eqv.), M2 (185 mg, 0.50 mmol, 25 eqv.), diethyl 2-bromo-2-methylmalonate (5.4 mg, 0.021 mmol, 1 eqv.), PMDTA (4.1 mg, 0.024 mmol, 1.1 eqv) and anisole (1.0 mL). The tube was sealed with a septum cap and the solution degassed by three freeze-pump-thaw cycles. The degassed solution was transferred to the sealed tube containing the copper (I) bromide; the resulting solution was stirred at 100 °C for several days and monitored with 1H NMR.
4.4.3 AIBN initiated polymerisations
A typical procedure for an AIBN initiated polymerisation of N-propyl maleimide and M2 was as follows. In a baked glass tube was added N-propyl maleimide (46.3 mg, 0.33 mmol, 50 eqv.), M2 (123 mg, 0.33 mmol, 50 eqv.), AIBN solution (33 μL, 0.2 M in toluene, 0.007 mmol, 1 eqv.) anisole (2.64 mL) and a magnetic stirrer bar. The tube was sealed with a septum cap and the solution degassed by three freeze-pump-thaw cycles. The degassed solution was stirred at 70 °C for several hours and monitored with 1H NMR.
4.4.4 Polymerisation monitoring
Monomer conversion was determined by 1H NMR by comparison of the integration of the broad polymer band (δ = 0.80 – 0.40 ppm) and the sharp triplet monomer peak (δ = 0.94 – 0.84 ppm) from the CH3 of the propyl group of N-propyl maleimide.
4.4.5 Polymer purification
All polymers were precipitated by adding the reaction mixture dropwise into the vortex of a vast excess of stirred methanol. The solid polymer was isolated by filtration (0.45 μm pore size filter paper) and dried under a vacuum.
4.5 Polymer hydrolysis
A typical procedure for the hydrolysis of a polymer in this project is as follows. To a stirred solution of polymer (40 mg) dissolved in THF (4 mL) at 40 °C, was added a solution of lithium hydroxide (36 mg, 20 equivalents per polymer repeating unit) in water (1 mL). The reaction was left for 16 hours, after which time the solvent was evaporated to give a crude containing the hydrolysed polymer, which was analysed without further purification.
Bibliography
1 W. A. Braunecker, Prog. Polym. Sci., 2007, 32, 93–146.
2 M. Ouchi, M. Nakano, T. Nakanishi and M. Sawamoto, Angew. Chemie – Int. Ed., 2016, 55, 1–7.
3 J. Preston, J. Polym. Sci. PART A-1, 1966, 4, 529–539.
4 R. M. Stayshich and T. Y. Meyer, J. Am. Chem. Soc., 2010, 132, 10920–10934.
5 A. M. Rosales, R. A. Segalman and R. N. Zuckermann, Soft Matter, 2013, 9, 8400–8414.
6 S. Srichan, N. Kayunkid, L. Oswald, B. Lotz and J.-F. Lutz, Macromolecules, 2014, 47, 1570–1577.
7 J. Li, S. N. Rothstein, S. R. Little, H. M. Edenborn and T. Y. Meyer, J. Am. Chem. Soc., 2012, 134, 16352–16359.
8 C. M. Dobson, A. Šali and M. Karplus, Angew. Chemie – Int. Ed., 1998.
9 K. A. Dill, Biochemistry, 1990, 29, 7133–7155.
10 J. Borg, M. H. Jensen, K. Sneppen and G. Tiana, Phys. Rev. Lett., 2001, 86, 1031–1033.
11 K. A. Dill, S. Bromberg, K. Yue, K. M. Fiebig, D. P. Yee, P. D. Thomas and H. S. Chan, Protein Sci., 1995, 4, 561–602.
12 A. R. Khokhlov and P. G. Khalatur, Phys. Rev. Lett., 1999, 82, 3456–3459.
13 M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917–924.
14 G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941.
15 J. F. Lutz, Macromolecules, 2015, 48, 4759–4767.
16 T. T. Trinh, L. Oswald, D. Chan-Seng and J. F. Lutz, Macromol. Rapid Commun., 2014, 35, 141–145.
17 T. T. Trinh, L. Oswald, D. Chan-Seng, L. Charles and J. F. Lutz, Chem, 2015, 21, 11961–11965.
18 R. K. Roy, A. Meszynska, C. Laure, L. Charles, C. Verchin and J.-F. Lutz, Nat. Commun., 2015, 6, 7237–7239.
19 U. S. Gunay, B. E. Petit, D. Karamessini, D. Gigmes, L. Charles and J.-F. O. Lutz, Chem, 2016, 1, 114–126.
20 H. Mutlu and J. F. Lutz, Angew. Chemie – Int. Ed., 2014.
21 V. Du Vigneaud, Science (80-. )., 1956, 123, 967–974.
22 R. B. Merrifield, Angew. Chem. Int. Ed. Engl., 1985, 24, 799–892.
23 J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–5.
24 Y. Hibi, M. Ouchi and M. Sawamoto, Nat. Commun., 2016, 7, 11064.
25 J.-F. Lutz, An Introduction to Sequence-Controlled, American Chemical Society, Washington DC, 2014.
26 M. Ueda, Prog. Polym. Sci., 1999, 24, 699–730.
27 J.-F. Lutz, Polym. Chem., 2010, 1, 55.
28 J.-J. Yan, D. Wang, D.-C. Wu and Y.-Z. You, Chem. Commun. Chem. Commun, 2013, 49, 6057–6059.
29 K. Satoh, S. Ozawa, M. Mizutani, K. Nagai and M. Kamigaito, Nat. Commun., 2010, 1, 6.
30 M. Krzysztof, Prog. Polym. Sci., 2005, 30, 858–875.
31 N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390.
32 J.-F. Lutz, T. Pakula and K. Matyjaszewski, in Advances in Controlled/Living Radical Polymerization, American Chemical Society, 2003, vol. 854, pp. 19–268.
33 P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794.
34 J.-S. Wang and K. Matyjaszewsk, Macromolecules, 1995, 28, 7901–7910.
35 V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165.
36 K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33.
37 J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024.
38 S. Srichan, University of Strasbourg, 2015.
39 M. Zamfir and J. F. Lutz, in ACS Symposium Series, American Chemical Society, Washington DC, 2012.
40 A. Haji-akbari, PNAS Early Addit., ?, 1–3.
41 V. Haridas and S. Naik, RSC Adv., 2013, 3, 14199–14218.
42 M. G. Baldwin, J. Polym. Sci. PART A, 1965, 3, 703–710.
43 G. Chen, Z. Wu, J. Wu, Z. Li and F. Li, Macromolecules, 2000, 33, 232–234.
44 J. M. G. Cowie, Ed., Alternating Copolymers, Plenum Press, New York, 1985.
45 J. F. Lutz, B. V. K. J. Schmidt and S. Pfeifer, Macromol. Rapid Commun., 2011, 32, 127–135.
46 E. Mishima and S. Yamago, Macromol. Rapid Commun., 2011, 32, 893–898.
47 Y. Maki, H. Mori and T. Endo, Macromolecules, 2008, 41, 8397–8404.
48 M. Hisano, K. Takeda, T. Takashima, Z. Jin, A. Shiibashi and A. Matsumoto, Macromolecules, 2013, 46, 7733–7744.
49 K. Satoh, M. Matsuda, K. Nagai and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 10003–10005.
50 N. ten Brummelhuis and M. T. Heilmann, Macromolecules, 2016, 49, 6879–6887.
51 G. B. Butler, J. Polym. Sci. Part A Polym. Chem., 2000, 38, 3451–3461.
52 Y. Hibi, S. Tokuoka, T. Terashima, M. Ouchi and M. Sawamoto, Polym. Chem., 2011, 2, 341–347.
53 Y. Hibi, M. Ouchi and M. Sawamoto, Angew. Chemie – Int. Ed., 2011.
54 W. Steglich and B. Neise, Angew. Chem. Int. Ed. Engl., 1978, 17, 522–524.
55 K. Duraimurugan, J. Sivamani, M. Sathiyaraj, V. Thiagarajan and A. Siva, J. Fluoresc., 2016, 26, 1211–1218.
56 W. Jakubowski, N. V Tsarevsky, P. McCarthy and K. Matyjaszewski, Mater. Matters, 2010, 5, 16–24.
57 K. Nishimori, M. Ouchi and M. Sawamoto, Macromol. Rapid Commun., 2016, 37, 1414–1420.
58 K. Matyjaszewski, N. V. Tsarevsky, W. A. Braunecker, H. Dong, J. Huang, W. Jakubowski, Y. Kwak, R. Nicolay, W. Tang and J. A. Yoon, Macromolecules, 2007, 40, 7795–7806.
59 J. Tom, B. Hornby, A. West, S. Harrisson and S. Perrier, Polym. Chem., 2010, 1, 420–422.
60 F. di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021.
61 K. Matyjaszewski, Macromol. Symp., 1998, 134, 105–118.
62 G. Liu, X. Li, L. Zhang, X. Qu, P. Liu, L. Yang and J. Gao, J. Appl. Polym. Sci., 2001, 83, 417–422.
63 G. Montaudo, F. Samperi and M. S. Montaudo, Prog. Polym. Sci., 2006, 31, 277–357.

[*] PMI = N-propylmaleimide, I = initiator = DEMM-Br, L = ligand = PMDETA
PMI = N-propylmaleimide, I = initiator = DEMM-Br, L = ligand = PMDETA
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Chemistry"
Chemistry is a science involving the study of the elements and matter at the atomic and molecular level including their composition, structure, properties, behaviour, and how they react or combine.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: