A Multi-step Synthesis of 4,5-Dimethylnonane
Info: 11279 words (45 pages) Dissertation
Published: 22nd Nov 2021
Tagged: Chemistry
Abstract
The work reported in this paper is an account of an attempt to synthesize the alkane 4,5-dimethylnonane via a multi-step reaction pathway. The process began with the Grignard addition of 2-bromopentane to hexan-2-one produce the tertiary alcohol 4,5-dimethylnonan-4-ol. The tertiary alcohol underwent a dehydration reaction under phosphorus oxychloride in pyridine to yield isomers of the alkene 4,5-dimethylnon-4-ene. Hydrogenation of the alkene over 10% Pd / C catalyst produced the target product.
4,5-dimethylnonane was successfully synthesized with a 3.9% yield and at 74% purity. Successful synthesis was confirmed with analyses by Fourier Transform infrared spectroscopy and gas chromatography, with both flame ion detection and mass spectrometry. The FT-IR spectrum showed characteristic alkane stretches, with strong C-H bending peaks 1458 cm-1, 1378 cm-1, and characteristic stretching and rocking at 1125 cm-1 and 739 cm-1 respectively. Purity was determined from proportions of GC-FID peak areas. GC-MS revealed a molecular ion peak at m/z = 156 with the base peak at m/z=57 and key losses at m/z = 70, 84, 99 and 113.
The Grignard reaction product was synthesised in a gross yield of 31.2 % while that of the POCl3 / pyridine dehydration was produced in a gross 32.6 % yield. These were insufficiently pure to characterise by GC-MS but assertions regarding their composition were made on the evidence of FT-IR and GC-FID analyses. The Grignard reaction rendered several undesired products: while exhibiting the -OH stretch characteristic of an alcohol at 3466 cm-1, carbonyl contamination was evidenced by a characteristic stretch at 1618 cm-1. Undesired side products were suggested to be products of enolisation, reduction, dehydration and Wurtz coupling reactions.
Carbonyl contamination of the dehydration product was successfully reduced by column chromatography. 4,5-dimethylnonan-4-ol was retrospectively identified by the absence of two GC-FID maxima following dehydration. The success of this step was indicated by the absence of the –OH stretch. The alkene isomers were not fully characterised: their presence was surmised from GC-FID peaks, but these were not individually attributed.
It was proposed that outcomes be improved by ameliorating the side reactions which occurred during Grignard addition by separating the products by chromatographic means or by addition of cerium (III) chloride. Following dehydration, analysis of the products by GC-MS following chiral column chromatography could confirm alkene isomerism and distribution. Similar methodology could also be applied to establish specific identification of the alkane isomers, alongside determination of enantiomeric excess by polarimetry.
Contents
1.0 INTRODUCTION 8
1.1 Grignard Addition to the Ketone 9
1.2 Alcohol Dehydration: Formation of Alkenes 14
1.3: Catalytic Hydrogenation of Alkenes 16
2.0 PROCEDURE 17
2.1 Grignard Addition to the Ketone 18
2.2 Alcohol Dehydration: Formation of Alkenes 20
2.3 Catalytic Hydrogenation of Alkenes 21
2.4 Analytical Techniques 22
2.4.1 FT-IR spectroscopy 22
2.4.2 Column chromatography 22
2.4.3 Gas chromatography with flame ionization (GC-FID) and mass spectrometric (GC-MS) detection 23
3.0 RESULTS 24
3.1 Grignard Addition to the Ketone 24
3.2 Alcohol Dehydration: Formation of Alkenes 26
3.3 Catalytic Hydrogenation of Alkenes 31
5.0 DISCUSSION 37
5.1 Grignard Addition to the Ketone 37
5.2 Alcohol Dehydration: Formation of Alkenes 39
5.3 Alcohol Dehydration: Formation of Alkenes 39
6.0 CONCLUSION 42
7.0 References 44
1.0 Introduction
The aim of this work was to synthesize the alkane 4,5-dimethylnonane via a multi-step reaction pathway. It was intended that chromatographic techniques including FT-IR, GC-FID and GC-MS be utilised to confirm the syntheses of expected products for each stage of the synthesis. The proposed reaction pathway is illustrated in Figure 1 below:
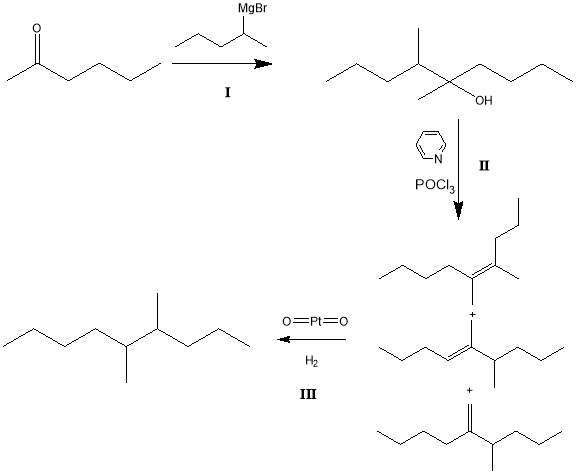
Figure 1: Proposed reaction pathway for the alkane synthesis
In Figure 1 the intended pathway is as follows:
I : Grignard addition of pentylmagnesium bromide to hexan-2-one in diethyl ether to produce the tertiary alcohol 4,5-dimethylnonan-5-ol.
II : Phosphorous oxychloride / pyridine dehydration of the alcohol to produce isomers of 4,5-dimethylnonene (not all conformers shown).
III : 10% Pd/C catalysed heterogeneous hydrogenation to synthesise 4,5-dimethylnonane.
1.1 Grignard Addition to the Ketone
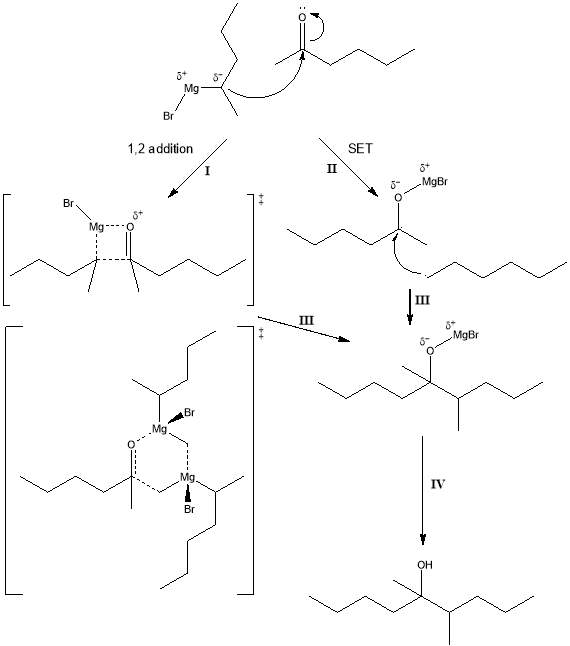
Figure 2: 1,2 addition and single electron transfer (SET) schemes proposed for Grignard reagent synthesis.
I : 4- and 6- constituent transition states.
II : Ketyl intermediate state.
III : Mg alkoxide.
IV: Tertiary alcohol.
The Grignard reaction is employed to yield alcohols- primary alcohols from formaldehyde, secondary alcohols from longer chain aldehydes, or tertiary alcohols from ketones. Other compounds may be prepared via Grignard synthesis, such as asymmetric ketones when reacting a Grignard reagent with a nitrile[1], or with carbon dioxide to synthesise a carboxylate[2]. Whatever the product, a Grignard reaction comprises two main steps: an organometallic halide is produced[3], and this subsequently undergoes addition to the carbonyl moiety of another molecule[4].
An organomagnesium halide, the Grignard reagent, is most commonly produced by the reaction of an alkyl- or aryl- halide with magnesium in a polar solvent, typically anhydrous diethyl ether or oxalane[5] (tetrahydrofuran). Aprotic solvents must be used in the reaction, since another solvent such as water or an alcohol will readily protonate the Grignard reagent to form a hydrocarbon. Further, the MgX bond is ionic and so the reaction requires that these bonds be solvated: in an ether the relatively polar C-O bond enables solvation and stabilising of the Mg ion by the oxygen component of the ether dipole[6]. Other methods such as dihalide conversion may also be employed to synthesise Grignard reagents[7], and other organometallic compounds such as organolithium reagents can be produced using similar methods to those described herein[8].
The Grignard reagent having been produced, its composition in solution is subject to changes dictated by the Schlenk Equilibrium[9]. In the Schlenk Equilibrium it is proposed that in diethyl ether, organomagnesium halides preferentially form complexes with two solvent molecules coordinated to the RMgX magnesium atom. Concentration of the solution, e.g. that caused by evaporation of the ether, will cause the equilibrium to favour the formation of MgX2 and R2Mg species[10], while concentrations of Grignard reagents greater than 0.1M result in complexation of the latter two species[6]. The Schlenk Equilibrium and those factors that affect its position are summarised in Figure 3.
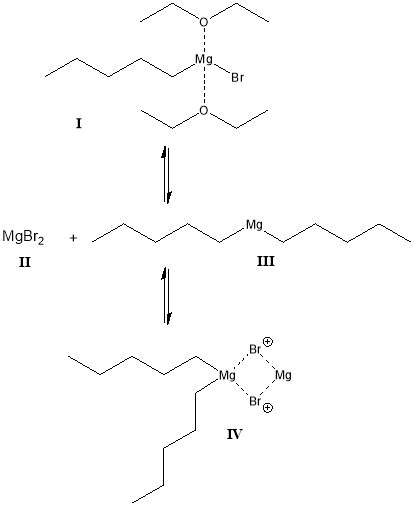
Figure 3: Schlenk Equilibrium of Grignard reagents in diethyl ether[5]
I : Complex formed in diethyl ether
II , III : Reduction in EtO2 volume shifts equilibrium to favour production of products
IV : Complex formed in significant amounts where the concentration of Grignard reagent is greater than 0.1M.
This variable speciation of organometallic species in ethereal solvents (Fig. 2) requires that the precise mechanism of the addition of an organomagnesium halide to a carbonyl compound remain a matter of discussion. One proposed mechanism describes the nucleophilic attack on the electrophilic carbonyl carbon by the organometallic α-carbon (with respect to the Mg) with the consequent formation of a four-membered transition state[11]. The resultant magnesium alkoxide species is then protonated either by a dilute strong acid such as H2SO4 or a weak acid solution such as a saturated NH4Cl solution to yield the alcohol product. An alternative mechanism wherein a six-membered transition state that involves two organomagnesium halide constituents has also been posited[12]. More recently, an altogether different mechanism involving single electron transfer (SET) from the organomagnesium halide to the carbonyl compound involving the formation of a ketyl intermediate has been proffered[13]. It was determined experimentally that the reagents undertook the SET pathway and that this became more apparent upon introduction a transition metal catalyst such as iron (III) chloride. The single ion transfer mechanism seems the more likely mechanism[12], as it also provides a mechanism by which aromatic and other sterically hindered ketones can undergo reaction.
Side products (Figure 4) that can form during the Grignard addition include production of an enolate species[14], a reduction product, a Wurtz-Grignard coupled species[15] and species formed by dehydration of the alcohol product. Enolation and reduction side reactions both involve the transfer of hydrogen. In enolation, hydride shift from the carbonyl constituent to the organomagnesium halide results in alkane formation. As a result, the carbonyl forms an enolate ion. This enolate ion then hydrolyses to reconstitute the carbonyl bond. In reduction, the transfer of a β-hydrogen from the organomagnesium halide to the carbonyl carbon results in the reduction of the alcohol, forming an alkene. These side reactions are observed more frequently where more sterically hindered reagents are used. Wurtz-Grignard coupling occurs during the SET reaction[16]: in the desired reaction, transfer of a halide ion to Mg·+ to produce the XMg· species is followed by the reaction of XMg· and R· species to form RMgX. Where random diffusion leads to two R· radicals meeting, R-R dimerization takes place. Tertiary alcohol products will readily undergo dehydration under ambient conditions to form alkenes as the stable tertiary carbocation, which serves as a reaction intermediate is formed.
Minimising losses to these side reactions has been investigated, and the use of cerium (III) halides[17], and of copper diphosphines[18] as catalysts has been proposed to this end. The issue of ambient dehydration, while not resolved, has been addressed by use during work up of a saturated solution of a weak acid such as NH4Cl rather than a solution of a strong acid[19].
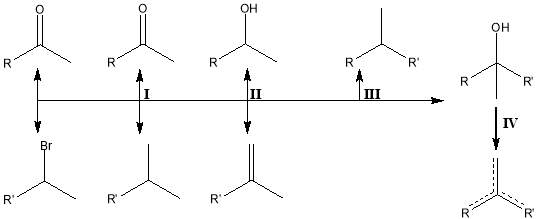
Figure 4: Side reactions and unwanted products formed during Grignard reaction.
I : Enolation and hydrolysis.
II : Reduction by the transfer of a β-hydrogen.
III : Wurtz-Grignard coupling.
IV: Ambient dehydration of the required product.
1.2 Alcohol Dehydration: Formation of Alkenes
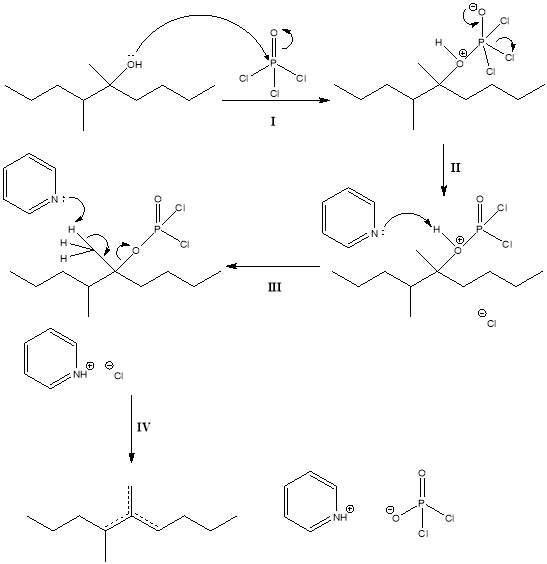
Figure 5: Mechanism for the phosphorus oxychloride / pyridine dehydration of 4,5-dimethylnonan-5-ol.
I : POCl3 reacts with tertiary alcohol to form the chlorophosphite leaving group
II : Elimination of a chloride.
III : Deprotonation by pyridine.
IV: E2 elimination to render the product.
Alcohol dehydration is a condensation reaction, proceeding by either the E2 pathway in the case of primary alcohols or the E1 pathway in the case of secondary or tertiary alcohols. Under acidic conditions, the nucleophile hydroxyl oxygen is protonated by the hydronium ion, to form an H2O leaving group. This leaves as water to render the carbocation intermediate. H2O then deprotonates the alkyl group nearest the positively charged carbocation of the intermediate, resulting in the formation of a double bond. Carbocation rearrangement occurs readily if an energetically favourable (i.e. more stable) conformation can be reached. It is for this reason of increased tertiary carbocation stability that tertiary alcohols readily dehydrate even at ambient temperature in only a mildly acidic environment[20, 21]. This can be ameliorated by using milder agents that are effective under milder conditions to undertake dehydration than monoprotic acids such as sulphuric acid[22]. It has been shown that pyridine solvated phosphorus oxychloride readily dehydrates tertiary alcohols. This reagent combination also finds use where a high degree of stereoselectivity is required, such as where a sterol substrate is undergoing dehydration[23]. Organosulphates such as toluenesulfonate or methanesulfonate can be used to perform a substitution reaction[24] by the hydroxyl on the sulphur, and then the sulphonate group may be eliminated, though this involves a two-step reaction. This regiospecificity becomes a consideration where dehydration of the alcohol will render isomers of the alkene product. Zaitsev’s rule[21] provides that the most stable product will be present in greatest abundance, with other enantiomers and diastereomers produced in smaller yields. Trans-substituted alkenes reduce steric hindrance by separating the substituents on each side of a double bond, and in more substituted alkenes, the π bond is stabilized by the adjacent alkyl substituents. The major product should therefore be the least sterically hindered trans-conformer of the most substituted alkene[25, 26].
There exists the potential for side reactions during this step. Where unreacted alcohol encounters a carbocation intermediate, it can react to form an ether[27]. Dimerisation / polymerisation can occur where the carbocation intermediate reacts with the product alkene[28]. By employing milder reagents such as pyridine / POCl3, no carbocation formation should take place, and the reaction should proceed via the E2 pathway[29]. The side reactions ought to be thereby eliminated. The use of a range metal species catalysts including copper sulphate and thorium oxide has also previously been studied experimentally but is not widely used[30].
1.3: Catalytic Hydrogenation of Alkenes
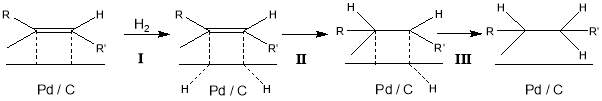
Figure 6: mechanism for the heterogeneous catalytic hydrogenation of a notional alkene using the Pd / C catalyst.
I : Adsorption of H ions to Pd / C substrate
II : Addition of H across the double bond yields alkyl radical
III : Addition of second H completes reaction, catalyst regenerated
The currently accepted theory for heterogeneous catalytic hydrogenation of alkenes proposes that firstly, the alkene and two hydrogen atoms adsorb to a metal catalyst surface[31]. There follows a sequential addition of a hydrogen atom across the double bond. The carbon-carbon double bond coordinates with surface of the catalyst from the least sterically hindered side first in a syn reaction. An example mechanism is provided in Figure 6.
Homogeneous catalysts that are miscible with the reaction mixture are widely used in hydrogenation and are the preferred catalysts for converting alkenes to their alkane derivatives[32]. Chiral phosphine complexes of noble metals (Ru, Rh, Ir etc.) are frequently selected and are known to provide high yields of alkanes with excellent enantioselectivity[33]. As well as this increased selectivity, homogeneous catalysts are less susceptible to catalytic poisoning[34] from agents including transition metals (e.g Al, Co) and TM oxides, sulphur- either as sulphide or thiol- which is present in laboratory rubberware, and amines. Heterogeneous catalysts which are dispersible in the reaction mixture are also used. These catalysts are distributed on catalytic supports such as carbon and silica, which serve to increase the available surface area as well as absorbing impurities[35]. A range of metals has been used in heterogeneous catalysis[36], with each offering different properties in terms of selectivity, temporal and / or financial cost and requisite reaction conditions, and so examination of the reaction requirements informs selection of the catalyst. Nickel (as Raney nickel) enables the selective reduction of conjugated systems at ambient conditions [37], while heterogeneous ruthenium catalysts hydrogenate conjugated systems under moderate conditions (70-80 °C, >50 atm)[38] with only partial selectivity. Rhodium catalysts offer greater versatility, able to reduce aromatic systems including heterocycles, carbon-carbon double bonds, carbonyls and nitro functional groups under mild conditions without acid. Each of these reductions can be carried out selectively by adjusting pH, and Rh catalysts are highly resistant to poisoning. This performance comes at a financial cost, with rhodium catalysts falling into the same price range as platinum catalysts. Other catalysts require vigorous reaction conditions- hydrogenation by copper chromium oxides occurs at temperatures of 150-220 °C and at pressures 100-150 atm[39].
Reaction conditions other than those required for effective catalyst activity are also to be considered. The substrate upon which the catalyst is to act may itself have reducible functional groups that require differing levels of work to hydrogenate, and hydrogenation selectivity can be modulated by pH and steric factors[40]. Selection of a solvent is therefore also reaction dependent [41]. Where adequate information regarding the effect of a solvent on a reaction cannot be obtained, a mixture of polar and non-polar solvents is used.
2.0 Procedure
Sections 2.1 – 2.3 describe the procedure carried out at each step of the synthesis. Before use, all glassware was thoroughly checked and found to be clean and free of defect before being placed in a drying oven at 40°C for 30 minutes. Section 2.4 describes instrumentation and settings. The molar quantities of reagents are dealt with in 4.0.
2.1 Grignard Addition to the Ketone
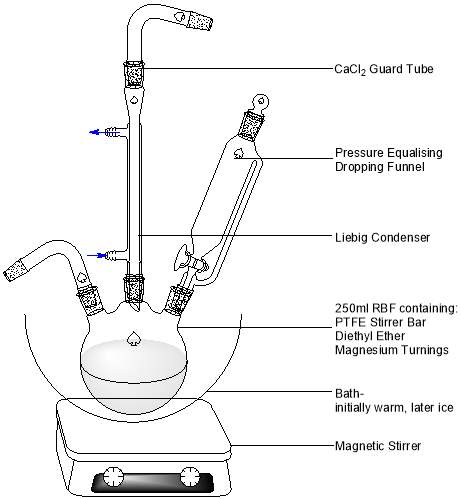
Figure 7: Apparatus used for Grignard reagent synthesis and addition to the ketone.
Oven-dried glassware was assembled without delay (Figure 8, above). To the three-necked RBF, 2.50 g of magnesium turnings and 40.0 ml of anhydrous diethyl ether (Na dried) were added, and magnetic stirring was initiated. 12.3 ml of 2-bromopentane were mixed with 10.0 ml of anhydrous diethyl ether and added to the pressure-equalizing dropping funnel. A warm water bath was introduced to initiate the reaction, and approximately 2 ml were quickly run in to the RBF. Subtle effervescence was observed, with a colour change from colourless to blue-grey. The remaining 2-bromopentane was added dropwise over a period of approximately 40 minutes, with constant gentle effervescence and a steady deepening of colour to a dark grey noted.
The reaction mixture was left to cool to ambient temperature (23°C) before being introduced to an ice bath for 10 minutes. 12.3 ml of hexan-2-one dissolved in 10.0 ml dry ether was added dropwise. A more vigorous effervescence was observed than that brought about in the previous step. The drop rate was adjusted so that that each drop was added as the effervescence of the last settled, taking approximately 20 minutes. When no further reaction was apparent, the ice bath was substituted for a warm water bath and a gentle reflux was initiated. This reflux was left to continue for 30 minutes, then removed to cool to ambient temperature.
The reaction mixture was transferred to a 250 ml beaker containing 50.0 g of crushed ice. 50.0 ml saturated aqueous ammonium chloride (27% w/v) solution was slowly added from a measuring cylinder in a constant and gentle stream, with constant stirring with a glass rod. Two layers formed: a colourless organic supernatant layer and a translucent white aqueous subnatant layer. The organic layer was run into a 250 ml QF conical flask from a 250 mL QF separating funnel. The aqueous layer was washed using three 20 ml aliquots of the ether before being discarded. The organic layer was dried under potassium carbonate in the stoppered 250 mL QF conical flask for 24 hours.
The dried organic layer was filtered under gravity using an 11 μm filter paper into a preweighed RBF before rotary evaporation. Rotary evaporation rendered a colourless oily liquid.
2.2 Alcohol Dehydration: Formation of Alkenes
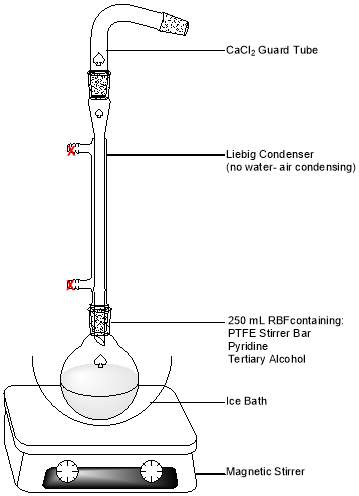
Figure 8: Apparatus setup for POCl3 dehydration of the alcohol product
Into a 250 mL round-bottom flask containing a PTFE stirrer bar, 12.0 ml of pyridine were measured, and 2.0 g of the previously prepared tertiary alcohol added. The flask was placed into an ice bath upon a magnetic stirrer, and stirring initiated. An air condenser topped with a CaCl2 guard tube was then attached. 8.0 ml phosphorus oxychloride (POCl3) were added dropwise with a Pasteur pipette. Dense white fumes were evolved, condensing to leave a chalky coating on the inside of the flask.
The reaction mixture was stirred for 24 hours. The resultant product was an oily orange liquid in which was suspended white particulate, of visibly lower volume than at the start. 35 g crushed ice was placed in a 250 ml separating funnel, and the contents of the flask carefully transferred by Pasteur pipette. The product was extracted three times with 15 ml aliquots of hexane and was observed to be colourless, the orange colour being confined to the discarded aqueous layer. The hexane layer was washed firstly with three 15 ml aliquots of 2M HCl and then by the same of distilled water. The hexane layer was dried for 24 hours using anhydrous sodium sulphate.
The dried organic layer was filtered under gravity using an 11 μm filter paper into a preweighed RBF before rotary evaporation. Rotary evaporation rendered a small quantity of a light straw coloured oil. Samples were taken for FT-IR and GC-FID analyses before column chromatography to investigate the product prior to hydrogenation.
2.3 Catalytic Hydrogenation of Alkenes
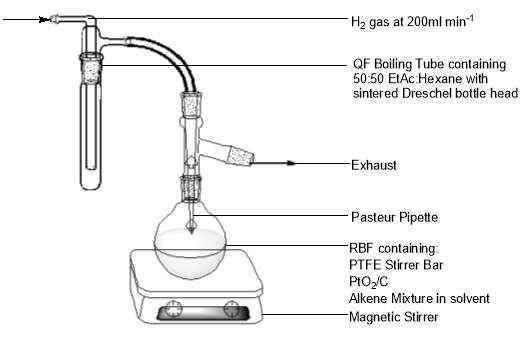
Figure 9: Apparatus used in the hydrogenation of alkenes to an alkane.
A 50 : 50 mixture of 100 ml hexane : ethyl acetate was prepared in a 250 ml RBF containing a PTFE stirrer bar using a measuring cylinder. 0.209 g of the alkene fractions were then added, followed by 200 mg of a heterogeneous 10% (m/m) palladium on carbon catalyst. A QF sidearm adapter with a secured Pasteur pipette was attached to the flask. A sintered Dreschel bottle head was attached to a QF boiling tube containing approximately 20 ml of the hexane/EtOAc solvent mixture. The Dreschel head was connected to the Pasteur pipette – sidearm adapter via rubber tubing. Magnetic stirring was initiated and the reaction mixture was hydrogenated at ambient conditions for a period of 8 hours using hydrogen gas at a flow rate of 150-200 ml min-1. The bulk of the mixture was then reduced by rotary evaporation.
When approximately 5ml of the mixture remained, filtration was undertaken through a slurry of 200 µm silica gel and 50:50 hexane : diethyl ether. Slurry was added to a cotton wool plugged Pasteur pipette to a depth of ≈1 cm. The filtrate was collected in a clean 7 ml glass vial and the solvent removed by nitrogen gas blowdown over 45 minutes. Samples were taken for FT-IR, GC-FID and GC MS analyses.
2.4 Preparation and purification
The methods used to purify or characterize the products of each synthesis step are outlined in the following sections, including instrumental parameters and general methods employed for sample preparation (GC-FID, GC-MS) and assembly of theapparatus for column chromatography apparatus.
2.4.1 FT-IR spectroscopy
Spectra were obtained in transmittance mode using a Brüker Alpha FT-IR spectrometer fitted with a Platinum ATR single reflection diamond ATR module. The diamond sample window was cleaned thoroughly with ethanol prior to background and in-between sample measurements. For analysis, two drops of sample were used and 16 scans carried out by the instrument. Spectra exhibited a resolution of 4 cm-1 and were obtained for the products of each synthesis step.
2.4.2 Column chromatography
A chromatography column was prepared with a 70-200 µm silica gel / hexane slurry stationary phase. The stationary phase was prepared by thorough mixing of 25.0 g silica gel with 50.0 ml of hexane in a beaker. This was transferred to the column and left to settle for 10 minutes. Excess hexane was run off, leaving a thin (
2.4.3 Gas chromatography with flame ionization (GC-FID) and mass spectrometric (GC-MS) detection
Analyses of products were undertaken using an Agilent Tech 7890A series gas chromatograph equipped with a 7683 series auto-sampler and a 7683B series autoinjector.
For GC-FID analysis, the gas chromatograph was fitted with an Rtx-5 (5%-Phenyl)-methylpolysiloxane column, 30 m x 0.32 mm ID, and a film thickness of 0.25 μm. Nitrogen was used as the carrier gas with a 1.0 ml min-1 flow rate. The injector volume was 1 μl and the injector temperature was 250 °C. The oven temperature was programmed to commence at 40 °C, rising by 10 °C min-1 to 300 °C, held at 300 °C for ten minutes. The flame ionization detector temperature was 300 °C, with hydrogen / air flow rates of 40 ml min-1 and 400 ml min-1 respectively.
For GC-MS analysis, the gas chromatograph was fitted with an Agilent HP-5ms column, 30 m x 0.25 mm ID, with a film thickness of 0.25 μm. Helium was used as the carrier gas with a 1.0 ml min-1 flow rate. The injector volume was 1 μl and the injection temperature was 300 °C. The oven temperature was programmed to commence at 40 °C, rising by 10 °C min-1 to 300 °C, held at 300 °C for ten minutes. The detector was an Agilent 5975A quadrupole mass selective detector with the ion source temperature set at 230 °C.
GC-FID samples were concentrated at ≈250 mg L-1 by serial dilution in hexane using 7 ml vials, with a 1.5ml aliquot of the sample transferred to a GC vial by syringe. GC-MS samples were prepared by dilution of their corresponding GC-FID samples by a factor that would render 100pA in GC-FID.
Mass Spectra were produced using OpenChrom Community Edition v1.1.0 (Diels)
3.0 Results
This section undertakes an examination of spectra and chromatograms obtained at each step of synthesis. With the exception of the alkane product, assignments are not absolutely certain. Yields and related calculations are dealt with in Section 4.
3.1 Grignard Addition to the Ketone
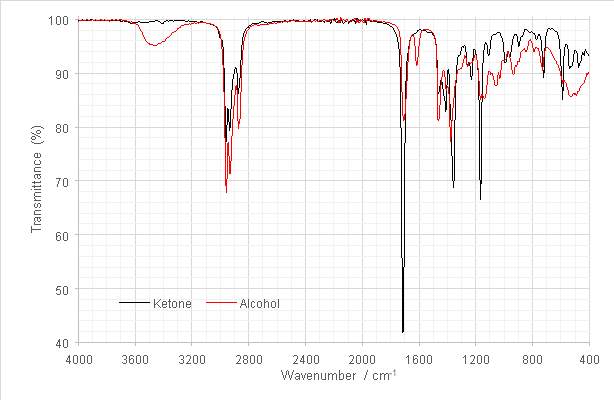
Figure 10: Overlaid FT-IR spectra of hexan-2-one and the tertiary alcohol product.
The product FT-IR spectrum obtained at this step exhibited the hydrogen bonded -OH stretch characteristic of alcohols between 3300-3600 cm-1. The greater intensity observed in the aliphatic C-H stretches between 2800 and -1 in the spectrum of the product were attributed to the addition of the pentyl group to the ketone during the Grignard reaction. The carbonyl stretch at around 1714 cm-1 was observed in both IR spectra, and so in the spectrum of the product was attributed to presence of ketone. This may have been unreacted reagent, or could have been formed partly or entirely by an enolisation side reaction, as posited in section 1.1 (Fig 4). The weak stretch at 1614cm-1 was attributed to a reduction side reaction resulting in a C=C species, also previously outlined in section 1.1 (Fig 4).
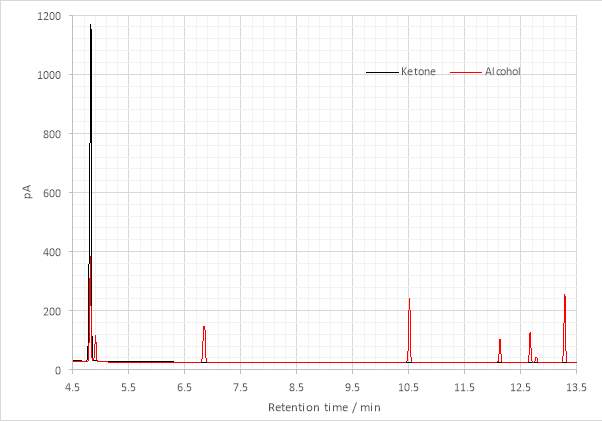
Figure 11: Overlaid gas chromatograms of hexan-2-one and the tertiary alcohol product.
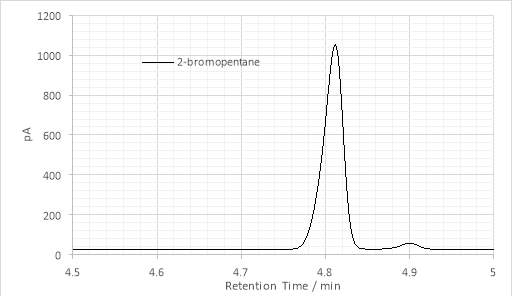
Figure 12: Single maximum GC chromatogram of 2-bromopentane
The persistence of ketone was confirmed by gas chromatography (Fig. 12). Also observable are several other peaks whose identities are not immediately clear, but whose presence was expected. The first, at ≈4.9 minutes was attributed to unreacted alkyl halide (Fig. 11). The maxima observed on GC-FID chromatograms of the Grignard product were not characterized by GC-MS as there was a mixture. However, the peaks could likely be attributed to the products of various side-reactions known to occur with Grignard addition to ketones, including enolisation, reduction, Wurtz coupling and tertiary alcohol dehydration processes.
3.2 Alcohol Dehydration: Formation of Alkenes
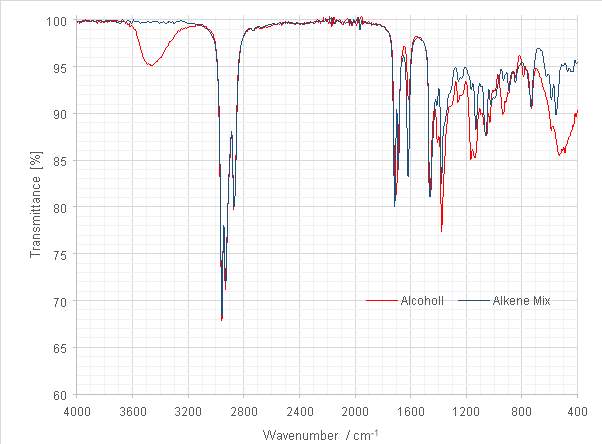
Figure 13: Overlaid spectra of the tertiary alcohol and dehydration product
The tertiary alcohol product was dehydrated overnight at room temperature (23 °C) using POCl3 / pyridine. The loss of the -OH stretch between 3300-3600cm-1 in the infra-red spectrum offered an initial indication of the successful conversion of the alcohol. The increase in intensity of the C=C stretch at 1618 cm-1 provided further indication of alkene formation. Carbonyl contamination was still evident, observed in the persistent C=O stretch at 1714 cm-1.
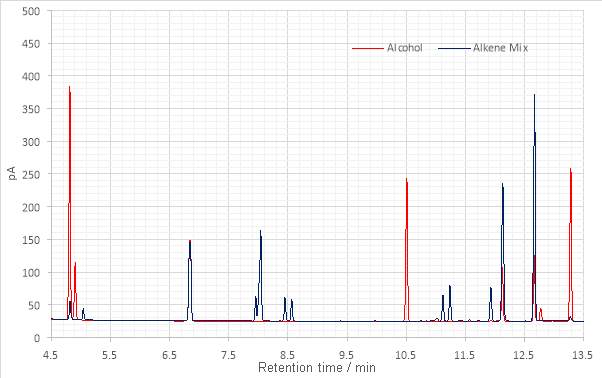
Figure 14: Overlaid gas chromatograms of the alcohol and dehydration products.
The major peaks present at 10.510 and 13.283 minutes in the alcohol chromatogram were not found in that of the dehydration product. Three other maxima, at 6.850, 12.133 and 12.670 minutes were visible on both. The maxima at 12.133 and 12.670 minutes exhibit an obvious increase in their peak areas.
Since the –OH stretch is absent from the FT-IR spectrum this was indicative that the maxima in the alcohol chromatogram at 10.510 and 13.283 minute might represent isomers of the chiral tertiary alcohol product. The peaks at 12.133 and 12.6704 were taken to be dehydration products, as their presence in the alcohol chromatogram was expected and has increased following dehydration. Seven new peaks- at 7.954, 8.034, 8.450, 8.563, 11.1217, 11.240 and 11.932 minutes were observed. Given that the target alkene has two stereocentres, isomerism in the alkene product was expected and therefore the appearance of new peaks was anticipated. Which were alkene and which were side products or contaminants could not be assigned with any degree of confidence.
The lack of change in the peak at 6.850, by inference of the identity of the other peaks, proposed that this might be an alkane product of Wurtz coupling.
The presence of carbonyl contamination evidenced by the FT-IR spectrum was to be addressed by means of column chromatography of the dehydration product. Following column chromatography, the first and fifth fractions evaporated completely during rotary evaporation. Fraction 2 was a small sample which vaporised in its flask shortly after it was sampled for FT-IR and was lost.
Table 1: Masses of fractions obtained by column chromatography.
| Fraction | Mass / g |
| 1 | 0 |
| 2 | 0.09 |
| 3 | 0.23 |
| 4 | 0.14 |
| 5 | 0 |
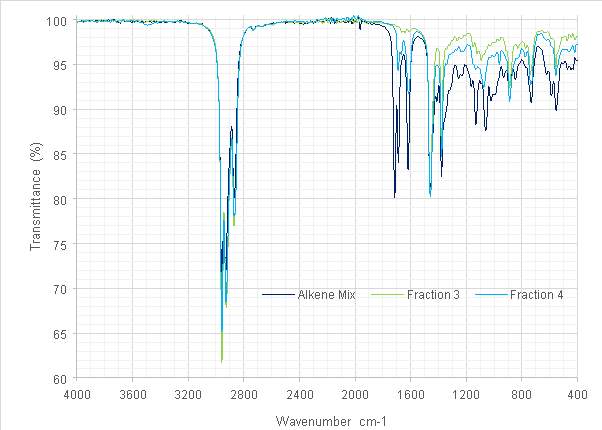
Figure 15: Overlaid spectra of the original alkene mix and two fractions.
The fractions recovered were analysed by FT-IR and GC-FID and compared (Fig 15). The reduction in intensity of the C=O stretch at 1714cm-1 in the alkene fraction IR spectra proposed a large reduction in carbonyl (and other) contaminants. While Fraction 2 was lost to evaporation, its spectrum (Fig. 16) proposes that it might have contained a disubstituted alkene, characterised by the 724 cm-1stretch. The reappearance of a small stretch at ≈3500 cm-1 in Fraction 4 was designated as possible contamination by water or by ethanol used to clean the diamond window of the instrument. Further investigation of the characteristic bands of either contaminant was not possible since at the intensities observed they would have been obscured by those of the product. Both fractions exhibited more definition in those peaks below 1000 cm-1 characteristic of alkenes.
The maxima previously suggested as the dehydration products of the alcohol were absent from the GC chromatograms produced for Fractions 3 and 4. Whether these did not elute during column chromatography, were lost during rotary evaporation or formed some part of Fraction 2 is not known. While this outcome was not desired, it did indicate that some contaminants might also have been removed. Of the peaks that were produced, the first at 6.850 min is still present from the previous step. Those at 7.954, 8.034, 8.450 and 8.563 were suggested to be alkene isomers.
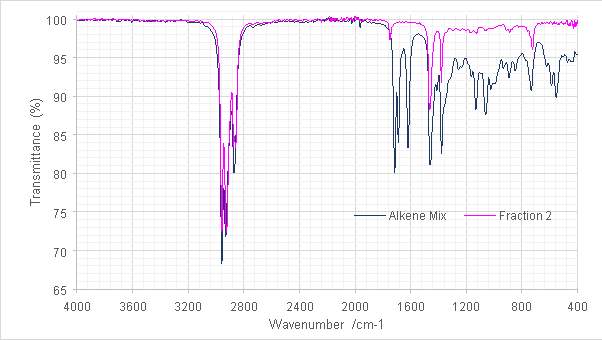
Figure 16: Overlaid spectra of the parent alkene mix and fraction 2.
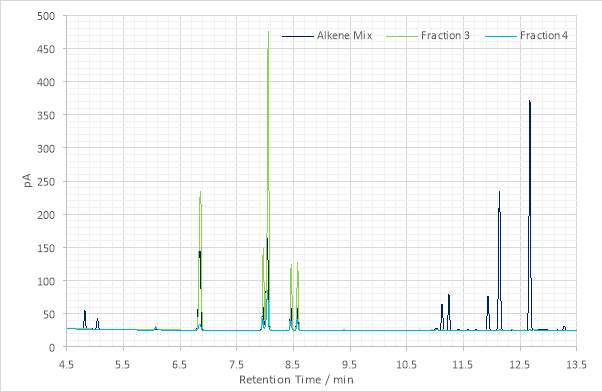
Figure 17: Overlaid gas chromatograms of the parent alkene mix, Fractions 3 and Fraction 4.
3.3 Catalytic Hydrogenation of Alkenes
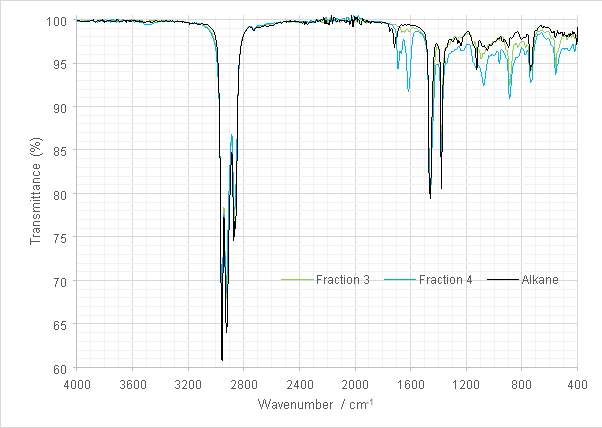
Figure 18: Overlaid spectra of the product alkane and the alkene fractions retained for hydrogenation.
It was decided to recombine fractions 3 and 4 as yields in previous steps had been low (see Section 4) and there was concern that isolating a final product may prove problematic. The FT-IR spectrum of the product displayed stretches characteristic of alkanes, with a –(CH2)n– rocking 739cm-1 and a stretch characteristic of branched alkanes at 1125 cm-1. C-H bending was exhibited at at 1458 and 1378 cm-1, and C-H stretching at 2800--1. The IR spectrum offered the first evidence of successful alkane formation.
The GC-FID chromatogram appeared to corroborate this finding, with the loss of all previous maxima (save the persistent peak at 6.850 min) and the appearance of a new pair of peaks at 8.208 and 8.243 min. The target alkane has two stereocentres and therefore co-eluting enantiomers and stereoisomers (SS’ and RR’, RS’ and SR’) were expected to be present as two peaks. Following consultation with Dr. L. Smik it was decided that the sample was acceptable to undergo characteristation by GC-MS analysis.
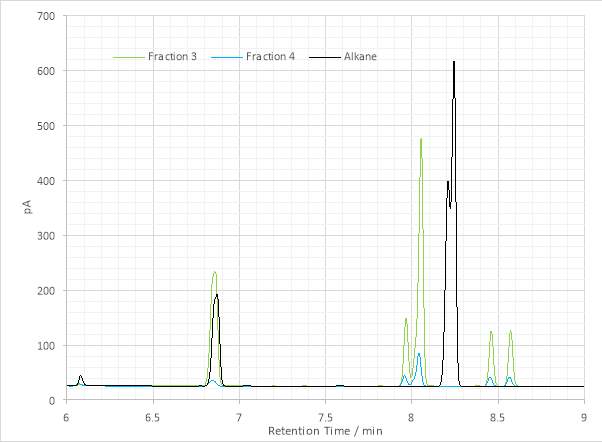
Figure 19: Overlaid gas chromatograms of the product alkane and the alkene fractions used for hydrogenation.
The molecular ion (M) was observed in the mass spectra of both peaks at m/z = 156 and corresponds to the relative molar mass of the target alkane, 156.19 g mol-1. The base peak was identified at m/z = 57 and was assigned as a C4H9· constituent that fragmented at a chiral carbon. Further assignation revealed that all of the main peaks fragmented at a stereocentre. The peak at m/z ≈ 84 and was assigned to the loss of the C6H13· fragment, while the peak at m/z ≈ 70 was explained by the loss of a C5H11· fragment. It was noted that these latter assignations are approximately 1 amu lower than the RMM of the fragments drawn below. This is due to the propensity of fragmenting molecules to lose hydrogen[1]. These and other assignments are summarised in Table 2.
These data, taken with the analyses of the FT-IR spectra and GC-FID chromatography enabled confident identification of the final product as the target alkane 4,5-dimethylnonane, present as two sets of enantiomers. This conclusion confirmed the previous interpretations of alkene (i.e. pre-hydrogenation product). The presence of three major maxima in the alkene chromatogram (Figure 30) and their subsequent collapse into one peak following hydrogenation was explained by formation of a new chiral carbon centre following the dissolution of the carbon-carbon double bond at the hydrogenation step.
Table 2: Assignment of the main fractions in Figures 21 and 22 with numbered reference to the main molecule.
| Fragment | Formula | Mass | 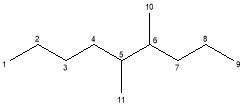 |
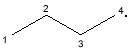 |
C4H9· | 57 | |
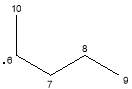 |
C5H11· | 70 | |
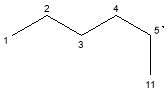 |
C6H13· | 84 | |
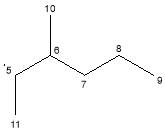 |
C7H15· | 99 | |
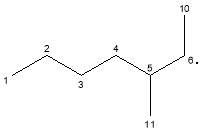 |
C8H17· | 113 |
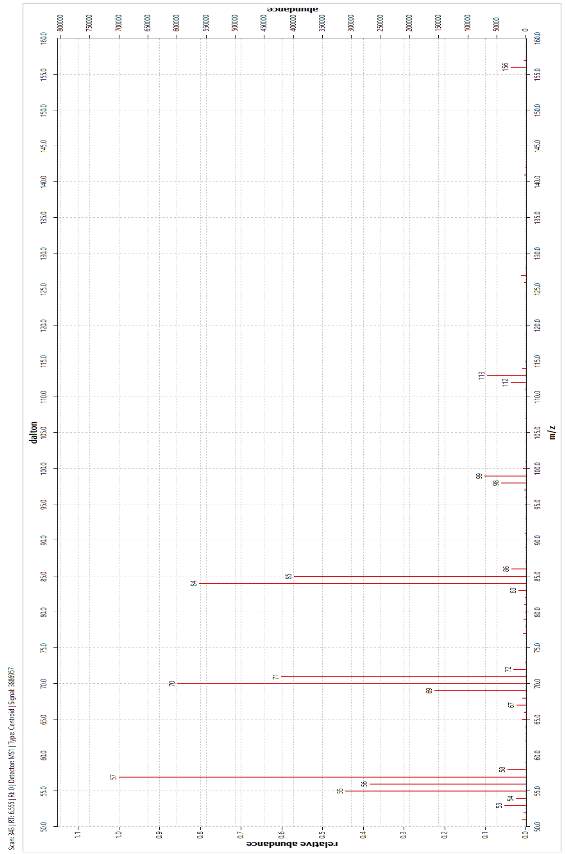
Figure 20: Mass Spectrum of one of the enantiomer pairs produced by the hydrogenation of alkenes (GC FID retention time 8.208 min)
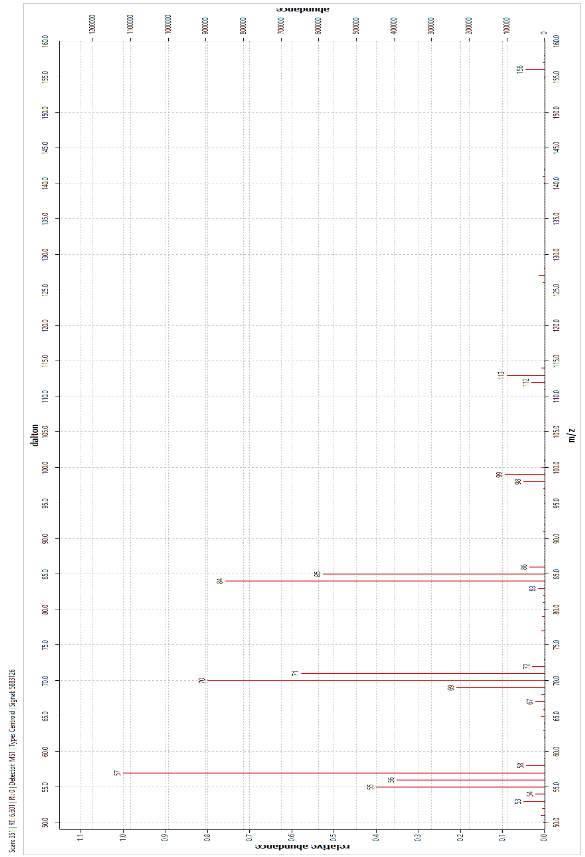
Figure 21: Mass Spectrum of one of the second set of enantiomer pairs produced by the hydrogenation of alkenes (GC FID retention 8.243 time min)
4.0 Calculation
The theoretical and percentage yields for each step were determined and tabulated. Yields were calculated under the following assumptions:
- That both the starting reagents and their products were pure.
- That the alkene was the limiting reagent for the alkane synthesis, as hydrogen was present in enormous excess.
- That the 10% Pd / C catalyst would emerge from the reaction unchanged and therefore its stoichiometry need not be considered.
For each step, the following process was applied:
V·d / RMM = n and/or
m / RMM = n
Stoichiometry was standardised to the lowest value of n, and the lowest value assigned as the limiting reagent (nLIM) in determining the theoretical and percentage yields:
nLIM · RMMPROD= yieldT
100 · (mPROD / yieldT) = % yield
Table 3: Yield calculation
| Hexan-2-one | Magnesium | 2-bromobutane | Product | |
| RMM / g mol-1 | 100.159 | 24.305 | 151.045 | 172.308 |
| d / g cm-3 | 0.8113 | – | 1.223 | – |
| V / ml | 12.3 | – | 12.3 | – |
| m / g | – | 2.5 | – | 5.36 |
| n / mol | 9.963×10-2 | 0.103 | 9.959×10-2 | – |
| Stoichiometry | 1.0004 | 1.03 | 1 | – |
| Theoretical yield / g | 17.16 | |||
| % yield | 31.23 |
Here, determining the limiting reagent was carried out as a matter of convention. Either reagent might have been used for calculation, with the difference of 4×10-5 mol making no difference to the theoretical yield value upon rounding. Densities of the reagents were taken from manufacturer data.
Table 4: Yield calculation for the dehydration step
| Alcohol | Pyridine | POCl3 | Product | |
| RMM / g mol-1 | 172.305 | 79.1 | 153.332 | 154.292 |
| m / g | 2.02 | 12 | 8 | 0.59 |
| n / mol | 0.012 | 0.152 | 0.052 | – |
| Stoichiometry | 1 | 12.941 | 4.450 | – |
| Theoretical yield / g | 1.81 | |||
| % yield | 32.62 |
Table 5: Yield calculation for the hydrogenation step
| Alkene | H2 | Product | |
| RMM / g mol-1 | 154.292 | 1.0079 | 142.282 |
| m / g | 0.209 | excess | 0.0729 |
| n / mol | 1.35×10-3 | excess | – |
| Theoretical yield / g | 0.192 | ||
| % yield | 37.82 |
The overall yield of the final alkane was calculated as a product of linear yields:
0.3123·0.3262·0.3782 = 3.85 %
The purity of the final product was calculated as a percentage of the peak area:
100·(AreaPROD / AreaTOTAL)
100·(3441.435 / 4663.354) = 73.8 %
5.0 Discussion
Discussion of each synthesis step is dealt with sequentially. The sections include a précis of the results, an investigation of the data regarding synthesis of the desired product, and suggestions regarding the experimental and analytical procedures that may offer more successful outcomes.
5.1 Grignard Addition to the Ketone
The appearance of the hydrogen bonded -OH stretch at 3443 cm-1 in the FT-IR spectrum Grignard reaction product offered an initial indication that the tertiary alcohol had been successfully produced, with the increase in intensity of the sp3 -CH stretch attributed to the addition of the pentyl group of 2-bromopentane to the ketone during the reaction. A C=O stretch at 1714 cm-1 indicated carbonyl contamination and the GC-FID chromatogram of the product revealed considerable contamination with side-products, with a total of 8 maxima observed.
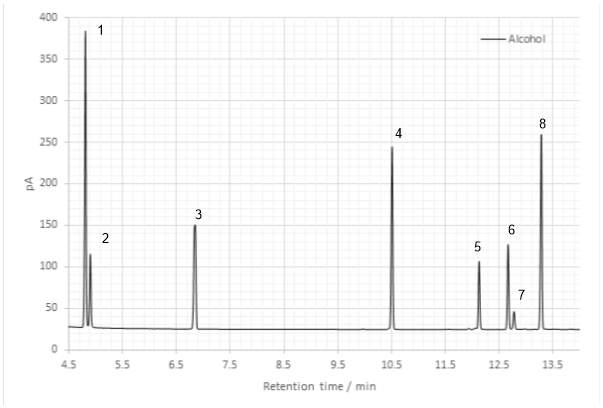
Figure 22: Gas chromatogram of the Grignard product
Peaks 1 and 2 are accounted for as unreacted ketone and 2-bromopentane respectively; the first is the only peak present in the GC chromatogram of the ketone and is present in that of the Grignard product, while peak 2 is the only on present in the 2-bromopentaner chromatogram. Two of the peaks, 4 and 8, were suggested as the tertiary alcohol product; the alcohol is a chiral molecule and therefore its stereoisomers elute at different times, though enantiomers co-elute. Both peaks were absent following POCl3 / pyridine dehydration. Peaks 5 and 6 may represent alkene isomers: they would have been present in the alcohol from dehydration, and their peak area increased greatly following dehydration. The minor peak numbered 7 could have contained an alcohol group, as it was absent following dehydration, and could have come about by an enolation or reduction side reaction. Peak 3 was persistent throughout, with no significant change in peak area. The final side reaction that commonly occurs during Grignard addition is the Wurtz coupling of excess alkylhalide and the Grignard reagent. During this reaction such a coupling would produce a C10 species. As this peak was present throughout, if it were an alkane it would have been unaffected by the subsequent reactions. Investigating the peak by GC-MS showed a molecular ion at m/z =142, with a base peak at m/z = 70 and other key losses at m/z = 57 and 99.
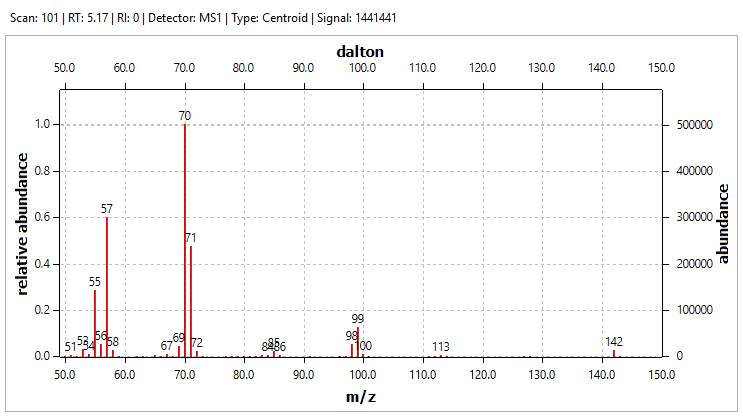
Figure 23: Mass spectrum of Peak 3.
Table 6: Fragments of 4,5-dimethyloctane
| Fragment | Formula | Mass | 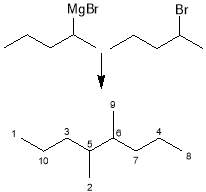 |
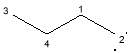 |
C4H82· | 57 | |
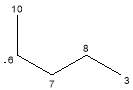 |
C5H11· | 70 | |
 |
C7H15· | 99 |
The possible fragmentation of the mass spectrum of a C10 product formed by Wurtz coupling is set out in Table 6. While this data and the persistence of the contaminant offered a plausible identification of peak 3, it is not possible to state confidently that this is the compound in question. Isolation by column chromatography and more rigorous interrogation of the data would be required.
Such questions might be avoided with changes to the experimental procedure. Cerium (III) halides have been shown to reduce side reaction products. The use of a copper diphosphine catalyst has also been reported to reduce side reactions in cyclic Griganard reactions.
5.2 Alcohol Dehydration: Formation of Alkenes
Initial indication of the successful dehydration of the Grignard product was evidenced by the FT-IR spectrum. This showed elimination of the –OH stretch and increased intensity of the C=C stretch at 1618 cm-1.
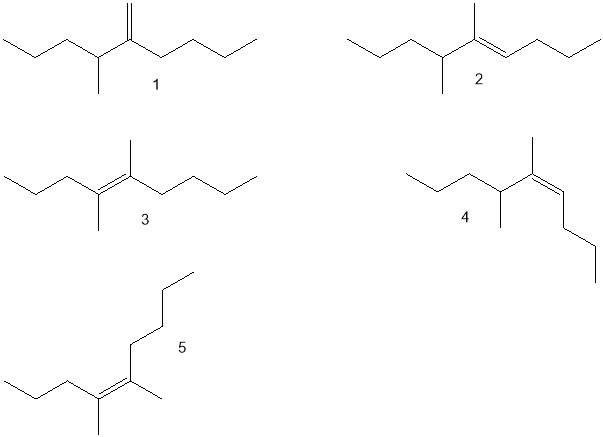
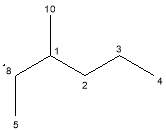
Figure 24: Alkene isomers- S and R conformations not shown.
Carbonyl contamination was still present, indicated by the carbonyl stretch at 1714 cm-1, although the carbonyl at peak 1 was greatly reduced, with no change to peaks 2 or 3. In Figure 22, peaks 5 and 6 were identified as dehydration product alkenes. In Figure 25 these maxima are seen as peaks 11 and 12 with greatly increased peak areas.
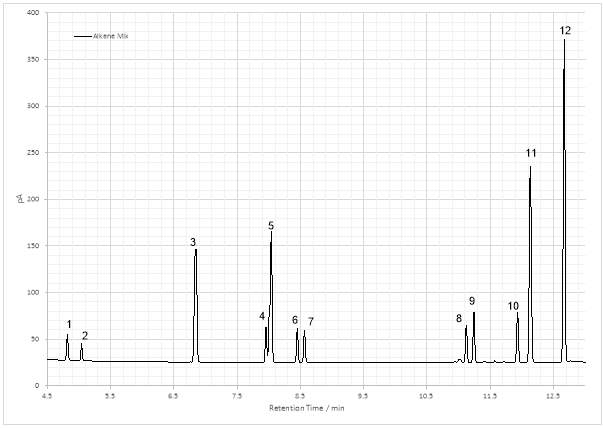
Figure 25: Gas chromatogram of the dehydration product
The identification of individual isomers from the GC chromatogram was problematic, since there were calculated to be five isomers (Fig 24) before the chiral centre was considered. Recalling Zaitsev’s rule and considering the maxima as indicative of abundance isomer, it was still possible to consider some possibilities regarding some of the peaks. The most substituted Z-isomers (4 and 5, Fig. 24), Z-5,6-dimethylnon-4-ene and Z-4,5-dimethylnon-4-ene would form in greatest quantity, and as such could have been formed at peak 5, 11 or 12, with Z-4,5-dimethylnon-4-ene being the more stable of the two. This is due to the functional groups attached to the C=C bond being less sterically hindered if situated on opposite sides. The E-isomers, E-5,6-dimethylnon-4-ene and E-4,5-dimethylnon-4-ene (2 and 3, Fig 24) would be produced in lower quantities, and would therefore be accounted for by lower peaks, perhaps 4 and 9. The least substituted isomer (1, Fig 24) would be present in lowest abundance and so might reasonably be found at peak 6, 7 or 8. Without no corroborative data this cannot be presented as a conclusion, and the issues of co-elution of enantiomers and identification of stereoisomers further complicate the matter, as there were already more peaks than basic isomers. Even with chiral column separation, and careful GC-MS could only be used to confirm the isomerism of the compounds represented by the maxima, as alkene π bond exists as a resonance structure, therefore certain identification may remain elusive.
Following column chromatography, 5 fractions were processed in order to reduce carbonyl contaminants and isolate the alkenes. Only two fractions were retained during the process, despite care having been taken during execution. The chromatography column was handled with care and the rotary evaporation was done slowly at 26 °C with only intermittent submersion of the RBF. This outcome could be improved by employing a full eleutropic series to obtain more fractions, and by use of nitrogen blowdown for evaporation. The loss of alkenes resulted in uncertainty that might have been avoided. This might also have provided fractions fit to analyse/ characterise further.
5.3 Catalytic Hydrogenation of Alkenes
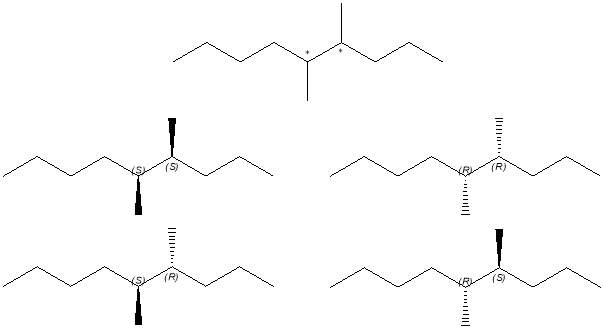
Figure 26: Isomers of the alkane,
As with the other syntheses, the IR spectrum offered the first evidence of successful product formation. The FT-IR spectrum of the product displayed stretches characteristic of alkanes, with a CH2 rocking peak at 739cm-1 and a stretch characteristic of branched alkanes at 1125 cm-1. C-H bending was exhibited at 1458 and 1378 cm-1, and C-H stretching at 2800--1.
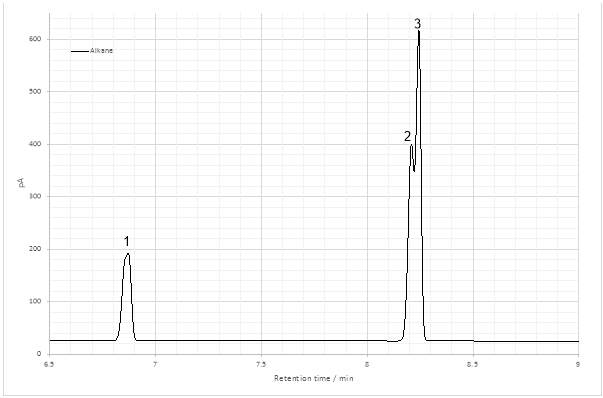
Figure 1: Gas chromatogram of the dehydration product.
The peaks of the gas chromatogram revealed the collapse of the alkene peaks (Fig 19) into a two very close peaks at 8.208 and 8.243 min. The structure contained two chiral carbons and therefore was likely formed by two sets of stereoisomers, the RR’ and SS’ enantiomers and the RS’ and SR’ enantiomers. Enantiomers co-elute and are not individually discernable in GC-FID, producing one peak. This is also generally true of GC-MS analysis.
The structure of the compound was confirmed to that of 4,5-dimethylnonene by GC-MS. The spectra (Figures 20 and 21) were in almost complete agreement. They both showed the molecular ion at m/z = 156, which was consistent with the molecular mass of the compound. The base peak was identified at m/z = 57 and was assigned as a C4H9· radical that fragmented at a chiral carbon. Full assignation of the main fragments amy be found in Table 2. The fragmentation pattern observed made it possible to confirm the structure being viewed. Taking the loss of the butyl radical at m/z = 57 as an example, the loss of a butyl radical from another part of the structure the structure would result in the formation of a secondary carbocation fragment. If the loss were a propyl radical, a primary carbocation would form. Each of these arrangements results in a less stable state.
While the target alkane was produced, further refinement can be applied to the process. Chiral chromatography could be used to separate the isomers, and examination under polarimetry could establish whether the enantiomers exist in differing proportions or as a racemate.
6.0 Conclusion
4,5-dimethylnonane was successfully synthesized in 3.85% yield via a multi-step reaction pathway. The structure was confirmed using a combination of FT-IR and GC-MS, and the purity of the compound was determined to be 74% by GC-FID. The product was most likely present as a mixture of RR’, SS’, RS’ and SR’ isomers given the presence of two stereocentres in the structure and the non-selective action of the Pd / C catalyst used in hydrogenation. This identification could be further refined by chiral HPLC to separate the isomers: use of polarimetry could determine the optical purity and enantiomeric excess of the constituents.
The products of the other reaction steps were identified with less specificity. Side reactions during the Grignard addition of 2-bromopentane to hexan-2-one produced a several undesired products due to dehydration, enolisation, Wurtz-Grignard coupling and reduction. Multiple peaks manifested during GC-FID chromatography whose identities were suggested (but could not be categorically stated) either retrospectively or by interrogation of FT-IR spectra and GC-FID chromatograms. Side reaction products could be separated by column chromatography using a suitable series of solvents, allowing characterization of products by more powerful analytical techniques such as GC-MS and NMR. The identity of the alcohol product was inferred by the disappearance of two peaks from the GC-FID chromatogram after the dehydration reaction. Generation of side products could be minimised or eliminated by addition of cerium (III) halide or copper diphosphine complexes during the reaction. POCl3 dehydration resulted in a mixture of alkene isomers which still contained the side-products from the Grignard addition step. Some alkene isomers were retained when attempting to separate these contaminants column chromatography. Characterisation of the alkene isomers was not possible by FT-IR and GC-FID: only the broadest suggestions could be made based on chromatogram peak areas recalling Zaitsev’s rule. With better separation, GC-MS could be used to characterise the products.
7.0 References
1. F. J. Weiberth and S. S. Hall. Copper(I)-activated addition of Grignard reagents to nitriles. Synthesis of ketimines, ketones, and amines. The Journal of Organic Chemistry, 1987, 52, 3901-3904.
2. B.-J. Li, L. Xu, Z.-H. Wu, B.-T. Guan, C.-L. Sun, B.-Q. Wang and Z.-J. Shi. Cross-Coupling of Alkenyl/Aryl Carboxylates with Grignard Reagent via Fe-Catalyzed C−O Bond Activation. Journal of the American Chemical Society, 2009, 131, 14656-14657.
3. H. M. Walborsky. Mechanism of Grignard reagent formation. The surface nature of the reaction. Accounts of Chemical Research, 1990, 23, 286-293.
4. F. Bickelhaupt. Organomagnesium chemistry: Nearly hundred years but still fascinating. Journal of Organometallic Chemistry, 1994, 475, 1-14.
5. F. W. Walker and E. Ashby. Composition of Grignard compounds. VI. Nature of association in tetrahydrofuran and diethyl ether solutions. Journal of the American Chemical Society, 1969, 91, 3845-3850.
6. E. C. Ashby, J. Laemmle and H. M. Neumann. Mechanisms of Grignard reagent addition to ketones. Accounts of Chemical Research, 1974, 7, 272-280.
7. H. J. R. de Boer, O. S. Akkerman and F. Bickelhaupt. An investigation of the di-Grignard approach to metallabenzocyclobutenes of group 14. Journal of Organometallic Chemistry, 1987, 321, 291-306.
8. J. J. Eisch. Henry Gilman: American Pioneer in the Rise of Organometallic Chemistry in Modern Science and Technology. Organometallics, 2002, 21, 5439-5463.
9. D. Seyferth. The Grignard Reagents. Organometallics, 2009, 6, 1598-1605.
10. J. Tammiku-Taul, P. Burk and A. Tuulmets. Theoretical Study of Magnesium Compounds: The Schlenk Equilibrium in the Gas Phase and in the Presence of Et2O and THF Molecules. The Journal of Physical Chemistry A, 2004, 108, 133-139.
11. E. C. Ashby and J. R. Bowers. Organometallic reaction mechanisms. 17. Nature of alkyl transfer in reactions of Grignard reagents with ketones. Evidence for radical intermediates in the formation of 1,2-addition product involving tertiary and primary Grignard reagents. Journal of the American Chemical Society, 1981, 103, 2242-2250.
12. K. Maruyama and T. Katagiri. Mechanism of the Grignard reaction. Journal of Physical Organic Chemistry, 1989, 2, 205-213.
13. T. Holm. Electron transfer from alkylmagnesium compounds to organic substrates. Acta chemica Scandinavica. Series B. Organic chemistry and biochemistry, 1983, 37, 567-584.
14. M. Hatano, O. Ito, S. Suzuki and K. Ishihara. Zinc(ii)-catalyzed Grignard additions to ketones with RMgBr and RMgI. Chemical Communications, 2010, 46, 2674-2676.
15. M. Anteunis and J. van Schoote. Studies on the Grignard Reaction-VII (1) on the Mechanism of the Grignard Reagent Formation and the Wurtz Side Reaction in Ether. Bulletin des Sociétés Chimiques Belges, 1963, 72, 787-796.
16. E. C. Ashby and J. Oswald. Concerning the mechanism of Grignard reagent formation. Evidence for radical escape and return to the surface of magnesium. The Journal of Organic Chemistry, 1988, 53, 6068-6076.
17. T. Imamoto, T. Hatajima, K. Ogata and M. Nishiura. Unusual cerium(III) chloride-promoted reactions of alkenyl grignard reagents or alkenyl-lithiums with 1,3-diphenyl-2-propanone: Formation and trapping of diorganometallic species. Applied Organometallic Chemistry, 1995, 9, 449-456.
18. A. V. R. Madduri, S. R. Harutyunyan and A. J. Minnaard. Asymmetric Copper-Catalyzed Addition of Grignard Reagents to Aryl Alkyl Ketones. Angewandte Chemie International Edition, 2012, 51, 3164-3167.
19. T. Poon, B. P. Mundy, J. McIntyre, L. Woods, F. G. Favaloro and C. A. Goudreau. Kinetic versus Thermodynamic Control in the Dehydration of 2-Methylcyclopentanol: A Two-Part Laboratory Experiment Utilizing the Gignard Reaction and GC-MS. Journal of Chemical Education, 1997, 74, 1218.
20. P. Vollhardt and N. E. Schore, Organic Chemistry: Structure and Function, Fifth edn., WH Freeman & Company, New York, USA, 2005.
21. J. Clayden, W. Warren, N. Greeves and P. Wothers, Organic Chemistry, Oxford University Press, Oxford, 2001.
22. J. S. Yadav and S. V. Mysorekar. A Facile Conversion of Tertiary Alcohols to Olefins. Synthetic Communications, 1989, 19, 1057-1060.
23. J. L. Giner, C. Margot and C. Djerassi. Stereospecificity and regiospecificity of the phosphorus oxychloride dehydration of sterol side chain alcohols. The Journal of Organic Chemistry, 1989, 54, 369-373.
24. A. I. Meyers and E. W. Collington. Facile and specific conversion of allylic alcohols to allylic chlorides without rearrangement. The Journal of Organic Chemistry, 1971, 36, 3044-3045.
25. A. Saytzeff. Zur Kenntniss der Reihenfolge der Analgerung und Ausscheidung der Jodwasserstoffelemente in organischen Verbindungen. Justus Liebigs Annalen der Chemie, 1875, 179, 296-301.
26. A. Saytzeff, Zur Kenntniss der Reihenfolge der Analgerung und Ausscheidung der Jodwasserstoffelemente in organischen Verbindungen, http://www.mjlphd.net/uploads/2/4/4/0/24404036/english_translation_of_1875_article_titled_the_order_of_addition_and_of_elimination_of_hydrogen_and_iodine_in_organic_compounds_by_alexander_saytzeff.pdf, Accessed 12th April, 2017.
27. B. C. Shi and B. H. Davis. Alcohol Dehydration: Mechanism of Ether Formation Using an Alumina Catalyst. Journal of Catalysis, 1995, 157, 359-367.
28. M. J. Janik, J. Macht, E. Iglesia and M. Neurock. Correlating Acid Properties and Catalytic Function: A First-Principles Analysis of Alcohol Dehydration Pathways on Polyoxometalates. The Journal of Physical Chemistry C, 2009, 113, 1872-1885.
29. W. J. Vera, M. S. Laya, P. S. Poon, A. K. Banerjee and E. V. Cabrera. Reagents for the synthesis of alkenes from carbonyl compounds: applications in the synthesis of terpenoid compounds. ARKIVOC, 2013, 1, 396-417.
30. A. B. Brown and E. E. Reid. The Catalytic Dehydration of Alcohols. The Journal of Physical Chemistry, 1923, 28, 1077-1081.
31. Z. N. Jaf, M. Altarawneh, H. A. Miran, Z.-T. Jiang and B. Z. Dlugogorski. Mechanisms governing selective hydrogenation of acetylene over [gamma]-Mo2N surfaces. Catalysis Science & Technology, 2017, 7, 943-960.
32. Y. Zhao, G. Fu and N. Zheng. Shaping the selectivity in heterogeneous hydrogenation by using molecular modification strategies: Experiment and theory. Catalysis Today, 2017, 279, Part 1, 36-44.
33. F. L. Lam, F. Y. Kwong and A. S. C. Chan, in Asymmetric Catalysis from a Chinese Perspective, ed. S. Ma, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 29-65.
34. A. J. Birch and K. A. M. Walker. Homogeneous hydrogenation in the presence of sulphur compounds. Tetrahedron Letters, 1967, 8, 1935-1936.
35. C. Gillan, Deactivation of precious metal steam reforming catalysts, PhD Thesis, University of Glasgow, 2010.
36. A. Wiersma, E. J. A. X. van de Sandt, M. A. den Hollander, H. van Bekkum, M. Makkee and J. A. Moulijn. Comparison of the Performance of Activated Carbon-Supported Noble Metal Catalysts in the Hydrogenolysis of CCl2F2. Journal of Catalysis, 1998, 177, 29-39.
37. A. F. Barrero, E. J. Alvarez-Manzaneda, R. Chahboun and R. Meneses. Raney Nickel: An Efficient Reagent to Achieve the Chemoselective Hydrogenation of α,β-Unsaturated Carbonyl Compounds. Synlett, 1999, 1999, 1663-1666.
38. T. Naota, H. Takaya and S.-I. Murahashi. Ruthenium-Catalyzed Reactions for Organic Synthesis. Chemical Reviews, 1998, 98, 2599-2660.
39. R. Prasad and P. Singh. Applications and Preparation Methods of Copper Chromite Catalysts: A Review. Bulletin of Chemical Reaction Engineering & Catalysis, 2011, 6, 63-113.
40. F. Joó, J. Kovács, A. C. Bényei and Á. Kathó. Solution pH: A Selectivity Switch in Aqueous Organometallic Catalysis—Hydrogenation of Unsaturated Aldehydes Catalyzed by Sulfonatophenylphosphane–Ru Complexes. Angewandte Chemie International Edition, 1998, 37, 969-970.
41. R. A. Rajadhyaksha and S. L. Karwa. Solvent effects in catalytic hydrogenation. Chemical Engineering Science, 1986, 41, 1765-1770.
42. G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Third edn., Wiley, New Jersey, USA, 2004.
43. D. H. Williams and I. Fleming, Spectroscopic Methods in Organic Chemistry, Fifth edn., McGraw Hill, New York, USA, 1995.
42. T. J. Adley, G. Matteo and R. T. B. Rye. The mass spectrum of 2-cyclohexenol: Loss of CH3⋅ and H⋅ from the molecular ion. Organic Mass Spectrometry, 1979, 14, 562-563.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Chemistry"
Chemistry is a science involving the study of the elements and matter at the atomic and molecular level including their composition, structure, properties, behaviour, and how they react or combine.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: