Endoplasmic Reticulum Stress Response and Alzheimer’s Disease
Info: 3065 words (12 pages) Dissertation
Published: 10th Dec 2019
Tagged: NeurologyAlzheimers
Endoplasmic Reticulum Stress Response and Alzheimer’s Disease
Abstract
The endoplasmic reticulum is a vital organelle found in eukaryotic cells. It performs many functions from protein synthesis and folding, to storing calcium ions and lipid biosynthesis. Under conditions where there is an accumulation of unfolded proteins, the endoplasmic reticulum is placed under stress and activates the unfolded protein response, which stops proteins from being translated, and can lead to cell apoptosis if not controlled. One disease where there is uncontrolled ER stress is the neurological disease Alzheimer’s. Three major characteristics of Alzheimer’s disease are the buildup of Amyloid-β senile plaques, hyperphosphorylated microtubule-associated protein tau in neurofibrillary tangles, and neuronal cell death. Understanding the pathways that each of these characteristics are related to the unfolded protein response may be critical to the treatment of Alzheimer’s disease.
1. Endoplasmic Reticulum and the Unfolded Protein Response
The endoplasmic reticulum (ER) is crucial to many functions in a eukaryotic cell. Specific functions include protein assembly and folding, disulfide bond formation, lipid biosynthesis, and calcium ion (Ca2+) storage [1].When functioning properly, correctly folded proteins are transported to the Golgi apparatus for distribution, while misfolded or unfolded proteins are broken down by the ER-associated degradation system [2]. This system is sometimes overwhelmed by an influx of unfolded/misfolded proteins due to certain pathological conditions such as oxidative stress [2]. This leads to an accumulation of unfolded proteins within the ER, leading to a condition known as ER stress [1].
This ER stress causes the unfolded protein response (UPR) system to be activated. This response serves to reduce the number of synthesized proteins that enter the ER through the use of three ER stress sensory proteins: protein kinase R-like endoplasmic reticulum kinase (PERK), the inositol-requiring enzyme 1 α (IRE1α), and activating transcription factor 6 (ATF6) [1]. ER stress, if not controlled, can lead to autophagy and eventually cell death due to apoptosis [1].
PERK is a type I transmembrane kinase in the ER that regulates the UPR system [3]. Under normal cell conditions, PERK is inactive due to the binding of GRP78. When ER stress occurs, GRP78 dissociates with PERK due to the presence of unfolded proteins, allowing PERK to self-phosphorylate and activate [3]. PERK then phosphorylates the eukaryotic translation initiation factor-2 (eIF2α), which reduces the influx of protein into the stressed ER [3] by inhibiting general mRNA translation [4].
IRE1α and ATF6 are also type I transmembrane kinases activated during ER stress. IRE1α dissociates with GRP78 so that it can bind to unfolded proteins and protect the cells from further damage [3]. IRE1α then catalyzes the splicing of X-box binding protein 1 (XBP1) mRNA, a potent transcriptional activator that regulates genes involved in ER protein synthesis, protein folding, and autophagy [4].
ATF6, in response to ER stress, moves to the Golgi complex, where it is cleaved before moving to the nucleus to activate the transcription of UPR genes [3]. The genes activated by the N-terminal of ATF6, p50ATF6, are chaperone proteins BiP/GRP78, transcription factors CHOP and XBP1, as well as other proteins [4].
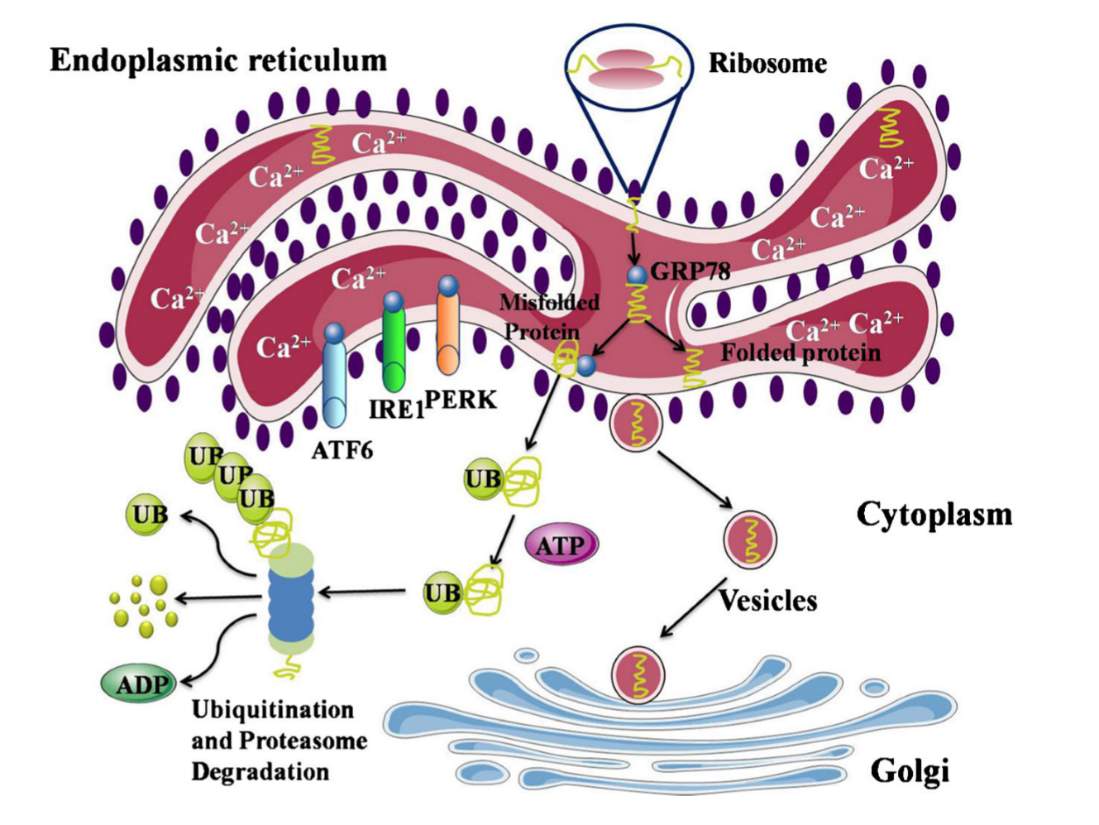
Figure 1: Normal state Endoplasmic Reticulum. Proteins created by the ribosomes are folded within the ER and are transported to the Golgi apparatus for distribution. GRP78 is bound to PERK, IRE1, and ATF6, causing them to be inactive. Misfolded proteins are degraded by the protein ubiquitin. Figure from [3].
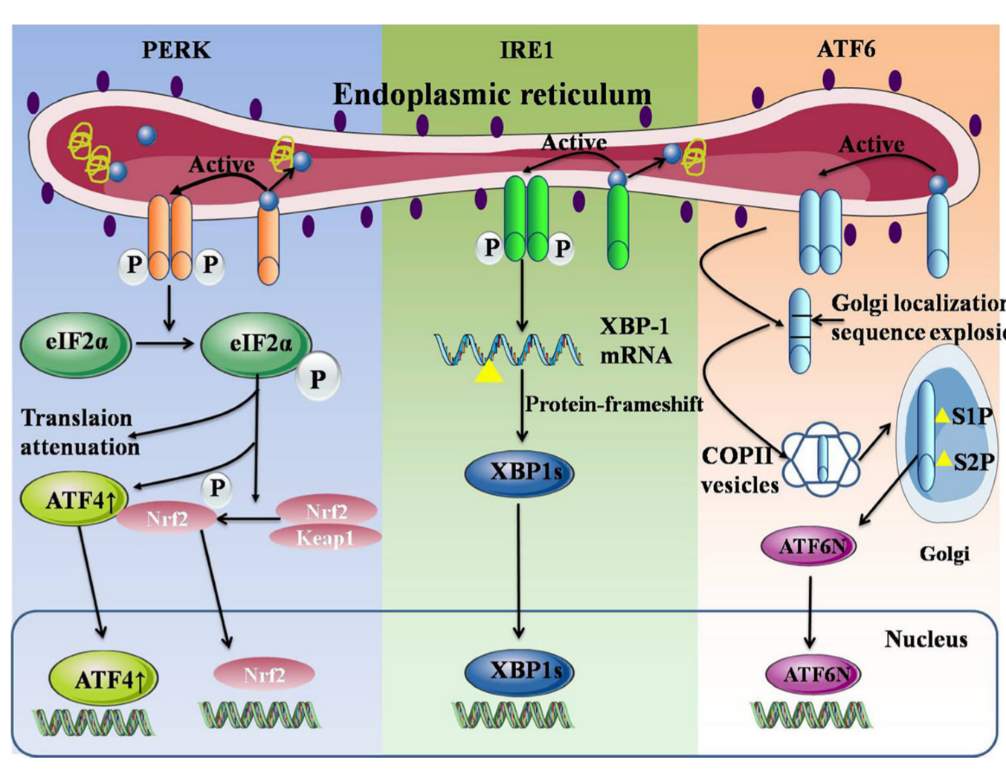
Figure 2: ER under UPR system. GRP78 unbinds from PERK, IRE1, and ATF6, activating their respective systems. Figure from [3].
1.1 Ubiquitin proteasome system and Autophagy
In order to degrade excess unfolded/misfolded proteins, a process called ER-associated degradation (ERAD) is activated in times of ER stress. Special channels in the ER export unfolded proteins to be degraded by the cytosolar ubiquitin proteasome system (UPS) [5]. This system is similar to autophagy, which is activated during ER stress. Changes to proteasome activity are identified in several neurological diseases such as Alzheimer’s disease and Parkinson’s disease [5].
Autophagy is the other major protein degradation system in cells and is activated during ER stress. When undergoing autophagy, membranes surround protein aggregates and the resulting vacuole fusses with a lysosome, forming an autophagolysosome, which degrades the protein aggregates [5].
2. Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive fatal neurodegenerative disease that results in a decline in memory and cognitive functions [6]. Major indicators of AD include the aggregation of Amyloid-β (Aβ) peptide in senile plaques, the accumulation of aggregated hyperphosphorylated microtubule-associated protein tau in neurofibrillary tangles, and synaptic and neuronal loss [7]. Thus, AD is representative of a protein-folding disease.
ER stress and UPR activation has been found to occur early on in neurofibrillary degeneration, and that the UPR is activated in the brain of a patient with AD [8]. Chronic ER stress caused by Aβ peptide accumulation activates a pro-apoptotic transcriptional factor CHOP, which suppresses the transcription of Bcl-2, an anti-apoptotic protein [9].
3. Amyloid-β
In the brain of a patient suffering from AD, amyloid-β (Aβ) peptides buildup on the outsides of neurons, called a senile plaque. Increased levels of Aβ induce chronic ER stress, which starts a pathway of oxidative stress, calcium ion deregulation, mitochondrial dysfunction, which can in turn further induce ER stress in a fatal cycle [10]. Aβ accumulation is linked to a disrupted cholesterol homeostasis, and patients with AD have been shown to have increased cholesterol levels [11]. Moderate levels of ER stress are known to deregulate lipid metabolism, and ER stress from Aβ buildup happens prior to cholesterol buildup in the mitochondria [11]. Aβ oligomers also induce Ca2+ dependent ER stress [12].
Aggregates such as Aβ can have a negative effect on proteasome complexes and function when studied in vitro [5]. In extreme cases such as those presented by AD, when the ERAD system is overwhelmed, and a large amount of unfolded protein might have aggregated within the ER and can’t be exported, the degradation of parts of the ER itself would be the more favorable pathway to try and restore homeostasis [5].
3.1 Humic Acid
Aβ deposits on neurons have been known to lead to cell death but the mechanism to cause apoptosis has been largely unknown. One link has been found to be between humic acids (HA), high molecular weight heteropolymers that are etiological factors for vascular diseases, and Aβ plaques [13]. When added to neural cells alone, HA produces no toxic effect, but when administered in the presence of Aβ plaque, apoptosis of the neural cells was increased than with the administration of Aβ alone or with a control acid, fulvic acid [13].
It was shown that HA, in the presence of Aβ, increased the phosphorylation of PERK and eIF2 , two of the indicators of ER stress [13]. The stress caused by HA and Aβ cause apoptosis by interfering with the expression of Sirt1 and PGC1 in mitochondria [13]. Sirt1, when overexpressed, managed to keep neural cells from dying due to the HA/Aβ stress [13].
3.2. HRD1, Syntaxin 5, Oxidative stress
One enzyme involved in the ER-associated degradation (ERAD) part of the UPR is the E3 ubiquitin ligase HRD1 [14]. This protein, expressed exclusively in neurons, plays an major role in the protection of cells against ER-stress induced apoptosis [14]. Under oxidative stress, HRD1 is rendered insoluble, which causes more proteins to build up and the cells to go to stress-induced apoptosis [14]. While a direct link between Aβ accumulation or hyperphosphorylated tau and HRD1 insolubility, Aβ generation may be promoted by the inhibition of HRD1, leading to further ER stress [14].
Oxidative stress also affects the expression of syntaxin 5, a vesicular transport receptor involved in membrane traffic, causing it to be overexpressed [15]. When overexpressed, syntaxin 5 causes the accumulation of Aβ peptides in the ER [15].
3.3 Resveratrol
An area of interest in treating AD is in polyphenols found in edible plants such as resveratrol, found in grapes, which slow down cognitive decline [16]. One of the targets identified was XBP1, which was necessary for resveratrol to reduce Aβ toxicity [16]. XBP1, which affects the transcription of genes that help fold proteins, is a part of the UPR, which is activated already in AD patients. Resveratrol also increased ubiquitin protein degradation activity [16].
3.4 Exercise
In an experiment in mice, it has been noted that exercise on a treadmill can alleviate some of the symptoms of AD [9]. After exercising with a treadmill, mice that had been mutated to have AD showed a reduced level of phosphorylation of PERK and eIF2α, critical components of the UPR [9]. Compared to sedentary mice in the same experiment, mice that were exercised showed suppression of pro-apoptotic pathways and a reduction in the expression of XBP1 and CHOP [9]. A deficiency of CHOP prevents ER stressed cells from continuing to stress-induced apoptosis [9].
4. Tau
Microtubule-associated protein tau is a protein that controls the assembly of the microtubule network component of a cell’s cytoskeleton [6]. The phosphorylation of the tau protei3n causes the protein to fall off of microtubules, causing them to fragment [6]. Hyperphosphorylated tau (P-tau) can form oligomers and toxic filaments known as neurofibrillary tangles (NFT) [17]. This buildup of tau filaments is a characteristic of AD, but the connection between ER stress and P-tau accumulation has been unknown [18].
One study proposed that tau activated the UPR system. Using a Tet-on controllable response element, tau expression was induced in cell lines to see if the ER stress of the cells was activated [19]. Two of the ER stress markers showed increased levels after the induced tau expression, indicating that tau itself started the initial accumulation of unfolded/misfolded proteins that starts the ER stress response [19].
4.1 HSF1
In non-neuronal cells, the heat shock response system helps to relieve ER stress [18]. A recent study has found out that in mice, the heat shock factor 1 (HSF1) decreased by 30% prior to the creation of hyperphosphorylated tau [18]. Once PERK (and the UPR) becomes activated, HSF1 levels fall even more to a 60% loss [18].
5. Rab6
Rab6 is a small GTPase involved in transport between the Golgi and ER [20]. The presence of Rab6 inhibits the production and formation of Aβ [20]. In the brain of an AD patient, the levels of Rab6 are increased by a factor of five when compared to cells that are not affected by AD [20]. The levels of Rab6 and BiP are correlated, which indicates that the amount of Rab6 parallels the extent of ER stress and suggests that Rab6 might be involved in the response to unfolded proteins [20].
One hypothesis put forward to the correlation between the two, since Rab6 transports proteins between the Golgi and ER, that upregulation of Rab6 could cause ER stress due to too many proteins being trafficked to the ER, causing the buildup that causes stress [20]. Another theory is that due to the presence of cytoskeletal abnormalities (NFTs) protein exit from the Golgi might be what is causing the accumulation of proteins that causes ER stress [20], as if the Golgi is full of proteins, then more cannot be transported from the ER, causing a buildup and stress.
So while Rab6 is correlated with the UPR, it is not elevated as a result of the activation of the UPR, but rather as a side mechanism to help relieve stress. If more transport proteins are made, then the ER could be trying to get the unfolded proteins removed before activating the UPR [20].
X. Conclusion
A fatal disease without a cure, Alzheimer’s disease affects nearly 5.5 million people in the United States. We have tried to understand its mechanisms, identifying plaques of amyloid-β peptide buildup on neural cells and mass tangles of hyperphosphorylated tau protein, to identifying how the cell’s own response to unfolded proteins can cause a cascade that ends in the cell’s premature death.
By understanding how the different characteristics of AD effect the cell’s response system, we can hope for a treatment or possibly even a cure. One promising area is exercise, which was shown to slow down the debilitating effects of AD in mice. By slowing down neuronal cell death, the disease might be able to be slowed. Another promising area when dealing with Aβ plaque buildup is the treatment with resveratrol, which was shown to target XBP1 to slow down apoptosis. While these treatments may look promising, most were performed within modified mice or in vitro, and not in living human patients, where the mechanisms might work differently.
Bibliography
1. Pereira, C.M.F., Crosstalk between Endoplasmic Reticulum Stress and Protein Misfolding in Neurodegenerative Diseases. ISRN Cell Biology, 2013. 2013: p. 22.
2. Liu, Z., et al., Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis, 2015. 6: p. e1822.
3. Li, J.Q., et al., Endoplasmic reticulum dysfunction in Alzheimer’s disease. Mol Neurobiol, 2015. 51(1): p. 383-95.
4. Bravo, R., et al., Endoplasmic Reticulum and the Unfolded Protein Response: Dynamics and Metabolic Integration. International review of cell and molecular biology, 2013. 301: p. 215-290.
5. Nijholt, D., et al., Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer’s disease. Cell Death and Differentiation, 2011. 18: p. 1071-1081.
6. Pająk, B., E. Kania, and A. Orzechowski, Killing Me Softly: Connotations to Unfolded Protein Response and Oxidative Stress in Alzheimer’s Disease. Oxid Med Cell Longev, 2016. 2016: p. 1805304.
7. Marwarha, G., et al., Gadd153 and NF-κB crosstalk regulates 27-hydroxycholesterol-induced increase in BACE1 and β-amyloid production in human neuroblastoma SH-SY5Y cells. Plos One, 2013. 8(8): p. e70773-e70773.
8. Hoozemans, J.J., et al., The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol, 2009. 174(4): p. 1241-51.
9. Kang, E.B., et al., Treadmill exercise represses neuronal cell death and inflammation during Aβ-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis, 2013. 18(11): p. 1332-47.
10. Plácido, A.I., et al., Enhanced amyloidogenic processing of amyloid precursor protein and cell death under prolonged endoplasmic reticulum stress in brain endothelial cells. Molecular Neurobiology, 2015. 51(2): p. 571-590.
11. Barbero-Camps, E., et al., Endoplasmic reticulum stress mediates amyloid β neurotoxicity via mitochondrial cholesterol trafficking. Am J Pathol, 2014. 184(7): p. 2066-81.
12. Alberdi, E., et al., Ca(2+) -dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell, 2013. 12(2): p. 292-302.
13. Li, H.H., et al., Humic Acid Increases Amyloid β-Induced Cytotoxicity by Induction of ER Stress in Human SK-N-MC Neuronal Cells. Int J Mol Sci, 2015. 16(5): p. 10426-42.
14. Saito, R., et al., Effects of oxidative stress on the solubility of HRD1, a ubiquitin ligase implicated in Alzheimer’s disease. PLoS One, 2014. 9(5): p. e94576.
15. Hernández-Zimbrón, L.F. and S. Rivas-Arancibia, Syntaxin 5 Overexpression and β-Amyloid 1-42 Accumulation in Endoplasmic Reticulum of Hippocampal Cells in Rat Brain Induced by Ozone Exposure. Biomed Res Int, 2016. 2016: p. 2125643.
16. Regitz, C., et al., Resveratrol reduces amyloid-beta (Aβ₁₋₄₂)-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans. Eur J Nutr, 2016. 55(2): p. 741-747.
17. Brunden, K.R., et al., Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: a focus on tau assembly inhibitors. Exp Neurol, 2010. 223(2): p. 304-10.
18. Kim, E., K. Sakata, and F.-F. Liao, Bidirectional interplay of HSF1 degradation and UPR activation promotes tau hyperphosphorylation. Plos Genetics, 2017. 13(7): p. e1006849-e1006849.
19. Abisambra, J.F., et al., Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci, 2013. 33(22): p. 9498-507.
20. Scheper, W., et al., Rab6 is increased in Alzheimer’s disease brain and correlates with endoplasmic reticulum stress. Neuropathol Appl Neurobiol, 2007. 33(5): p. 523-32.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Alzheimers"
Alzheimer’s disease is a neurodegenerative disease that causes a progressive decline in brain function, affecting memory, language, orientation, and reasoning. Symptoms usually begin mild and get progressively worse.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: