Abnormalities of Glucagon Secreting Alpha Cells: Development of Type 1 and Type 2 Diabetes Mellitus
Info: 5932 words (24 pages) Dissertation
Published: 16th Feb 2022
An Investigation on the abnormalities of glucagon secreting alpha cells, focusing on how these aid in the development of type 1 and type 2 diabetes mellitus; and how glucagon secretion can be altered and used therapeutically in the treatment of type of 2 diabetes mellitus.
Introduction
The pancreas is a rectoperineal organ located in deep abdominal wall, formed from the fusion of the dorsal and ventral buds in the foregut during embryotic development. The structural anatomy of the pancreas can split into four main parts: The head, neck, body and tail; although there are no cuts or borders that separate these parts, they help to understand how the pancreas is positioned in relation to the other organs in the abdomen and deep abdominal wall. The head is an extension of the uncinate process and curves, hooking beneath the superior mesenteric artery, the head lays in the arch of the duodenum. The body of the pancreas is below the stomach, crossing over the abdominal inferior abdominal aorta, superior mesenteric artery, and vein. The position of the neck overlays the superior mesenteric artery, with the tail hidden in the hilum of the spleen. (El-Gohary1 and Gittes2, 2018). Blood supply to the pancreas is via branches from the coeliac trunk known as the superior and inferior pancreaticoduodenal arteries whilst Venous drainage is via the superior hepatic portal vein.
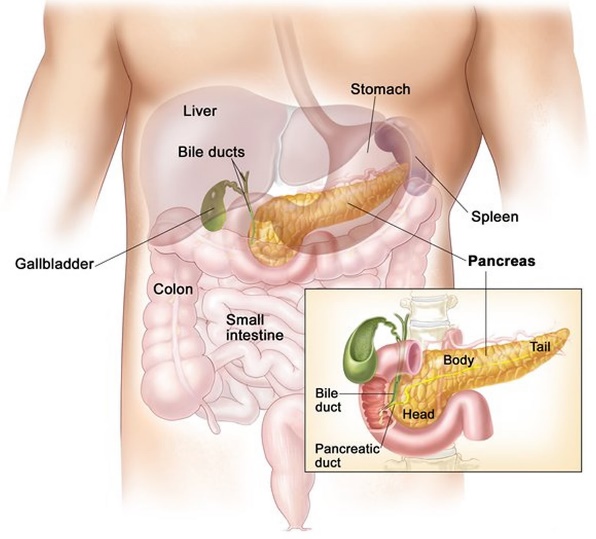
Figure 1: Anatomical position of pancreas showing the Head, Neck, Body and tail of the endo and exocrine organ in relation to the liver, curvature of the duodenum, spleen and gall bladder.
Image from (“Pancreas Location, Anatomy and Function in Digestion,” n.d.)
The pancreas is classified as both an exocrine and endocrine organ; its main endocrine function originating from the secretions of the α, β, δ, ε and PP cells in the islet of Langerhans. These cells control the secretions of insulin (controlled by the α-cells), glucagon (innovated by the β-cells) somatostatin and ghrelin (El-Gohary1 and Gittes2, 2018).
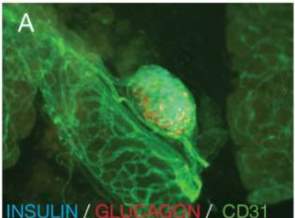
Figure 2: Image (A) shows mouse pancreatic tissue in helping to demonstrate the microcapillary network of islets of Langerhans with glucagon and insulin (El-Gohary1 and Gittes2, 2018).
Biochemical synthesis of Insulin
Insulin is a derivation of the preprohormone Preproinsulin which forms from the translation of the mRNA of insulin from the INS gene found in the p13 region of chromosome 11. Once the mRNA molecule has been translated, Preproinsulin is formed and acts as a precursor for proinsulin which forms from the removal of the signal peptide during insertion into the endoplasmic reticulum.
Proinsulin is comprised of three domains: a carboxy-terminal A chain, an amino-terminal B chain and a C peptide.
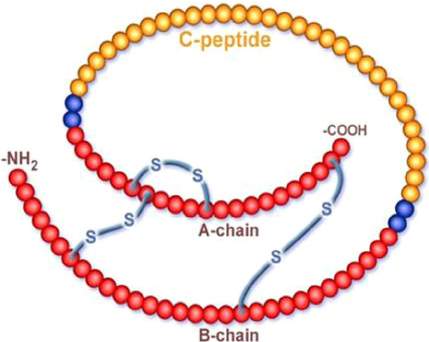
Figure 3: The structure of insulin showing the carboxylterminal A chain, amino-terminal B chain and C peptide.
Image from Serum C-peptide assay of patients with hyperglycaemic emergencies at the Lagos State University Teaching Hospital (LASUTH), Ikeja,” n.d.)
Upon exposure of β-cell endopeptidases, the proinsulin will separate (due to C-peptide excitation) and this unbound C-peptide and the now mature insulin (formed from the bonding of the a and b chain), will be packed by the Golgi apparatus into secretory vesicles. Once a signal has been received by the β cells in the islets as a result of increase plasma glucose concentration; the insulin will be secreted out of the cell via exocytosis of the vesicles, this will be diffused into the islet capillaries and then the blood. The free C-peptide is also secreted with the insulin but is thought to have no biological activity (“Insulin Synthesis and Secretion,” n.d.).
Control of insulin secretion
Glucose enters the β – cell via the GLUT2 glucose transporter, this is dependent on the elevated concentration of glucose in the extracellular fluid. Increased concentration of glucose leads to membrane depolarization of the β-cell innovating calcium influx from the calcium ion channels, this is thought to be a primary cause of exocytosis of insulin containing vesicles. Another pathway of activation of insulin secretion is hypothesized to be the unbalanced ATP to ADP concentration ratio (SGLT’s channels require ATP phosphorylation for the absorption of glucose in the enterocytes of intestinal epithelium (on luminal side) in the small intestine).
Biosynthesis of Glucagon
The gene responsible to glucagon synthesis encodes for the larger preprohormone proglucagon, this cleavage based on tissue specificity and goes on to become glucagon, GLP-1, GLP-2, oxyntomodulin and glicentin. Glucagon in a 29 amino acid peptide that once cleaved is secreted by the α cells of the islets of Langerhans. Glucagon has several receptors around the body but is generally associated with its action in the regulation (and opposition) of the actions of insulin, predominantly in the liver, and how this control promotes the pathways of gluconeogenesis and glycogenolysis whilst simultaneously decreasing glycogenesis.
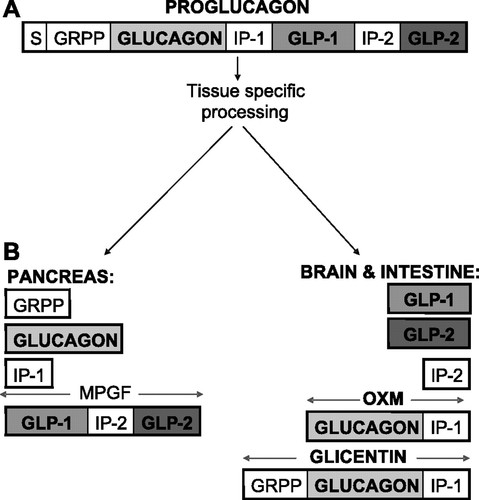
Figure 4:
A: structures of proglucagon B: tissue-specific posttranslational processing of proglucagon in the pancreas gives glicentin-related polypeptide (GRPP), glucagon, intervening peptide-1 (IP-1), and major proglucagon fragment (MPGF), whereas glicentin, oxyntomodulin (OXM), intervening peptide (IP-2), glucagon-like peptide (GLP)-1, and GLP-2 are liberated from the brain and the intestine. S, signal peptide.
GLP-1 is synthesized in the endocrine intestinal cells and has two main isoforms GLP-1(7-36) amide and GLP-1(7-37). GLP-2 is a 33 amino acid peptide that is secreted with GLP-1 in the intestinal endocrine cells in both the large and small intestine, where it acts upon; along with having receptors on the brain and adipose tissue, identified through cloning experiments that demonstrated the importance of the N-terminus in the bioactivity of GLP-1.
Receptor mechanism of glucagon
The actions of glucagon and peptides alike that are cleavage form the preprohormone proglucagon; actions are based on the G protein coupled receptor pathway. GLP-1 and GLP-2 active the Gαs pathway activating the target enzymes Adenylyl cyclase ATP converted to cAMP increase in secondary messenger cAMP activates protein kinase A and phosphorylates CREB protein which controls transcription of these genes. There is also action on the Gαq/11 activating phospholipase C which cleaves PIP2 to inositol triphosphate and DAG; this action of DAG actives protein kinase C, whilst inositol triphosphate acts as a secondary messenger in the endoplasmic reticulum binding to calcium ion channels and allowing the entry of calcium into the cell cytosol.
GLP-1 Receptor and action
Mice studies with a targeted mutation in the GLP-1 receptor ( through the ‘knockout’ of the germline inactivation of the GLP-1 receptor) showed that there was a mild glucose intolerance, this has allowed the further exploration into the action of GLP-1 in the islet, periphery and brain; as well as the GLP-R signalling in the islet with regards to its growth and proliferation (Lamont et al., 2012).
This mutation experiment can be applied when considering the polymorphisms that occur in the human genome, altering the activity of the GLP-1 receptor and other glucagon receptors alike. The GLP-1 receptor is located on chromosome 17q25, gene linkage analysis between families with a missense mutation in this chromosomal sequence, show an increased risk of developing type 2 diabetes due to a lower glucose response to glucagon; this is especially prevalent in the mutation of the Gly40Ser gene. These findings suggest that the Gly40Ser gene is involved in reducing blood glucose levels combatting hyperglycaemia (Sipos et al., 2015)
GLP-2 receptor and pathway of action
GLP-2 is an isoform of GLP-1, with the present of around 50% of the same amino acid sequence. The main difference between the two isoforms is that the gene that codes for the GLP-2 receptor is found in a different locus of chromosome 17 (17p13.3), unlike with the GLP receptor; there is no known linkage between human disease and this region of the chromosome.
GLP-2 follows the same pathway of active as the other forms of glucagon (Gαs and Gαq/11 pathway); but has less of an effect on the activation of MAP kinases (GLP-2 is independent of PKA activation despite its stimulation of the G-alpha s pathway.
Oxyntomodulin
Oxyntomodulin is another peptide compound that is cleaved from proglucagon; it is a 37 amino acid peptide, that contains all the 29 amino acids present in glucagon with the addition of 8 carboxyterminal extensions.
Its pathway of activation is not fully known, because whilst having no known receptor, it is hypothesized that it functions as a dual GLP-1 and GCGR agonist able to bind to both these receptors and cause activation, thus leading to the same effects that these molecules induce in tissue. Oxyntomodulin acts almost as an alternative molecule to GLP-1 and glucagon in low concentration; but it is unknown if this is in conjunction with other signalling molecules or independently of them.
Glicentin
Glicentin is another amino acid peptide cleaved from the proglucagon sequence (due to posttranslational processing in the intestine) and consists of a 69 amino acid sequence and an N-terminus. Glicentin is comprised of the glucagon 29 amino acid sequence, as well as a carboxyterminal extension and an N-terminal extension resulting in the 69 amino acid sequence of Glicentin. Biologically, glicentin is known to stimulate the secretion of insulin and the inhibition of gastric acid secretion which will promote gut motility and growth; as well as aiding in the restoration of normoglycemia; essentially, glicentin induces the same responsive pathway as glucagon, GLP-1 and GLP-2 in the body. At present it is unclear if high doses of glicentin can be used to subsidise weak activation of GLP and glucagon receptors in the body.
Glucagon receptor
The 29 amino acid peptide glucagon mediates its signalling effects on various biological systems in the body by binding to its receptor GCGR (Authier and Desbuquois, 2008). The GCGR follows the same action pathways GLP-1, GLP-2, oxyntomodulin and Glicentin (Gαs and Gαq/11) because they all belong to the family B receptors within the G protein coupled superfamily of seven transmembrane-spanning receptors. (Authier and Desbuquois, 2008).
Electrochemical control of glucagon secretion
The control of glucagon secretion is dependent on an increase or decrease in intracellular glucose secretion; as well as interactions between the secretion of insulin by the beta cells and somatostatin by the delta cell in the islets of the pancreas. The focus here will be the control of glucagon secretion through electrical regulation.
Alpha cells have multiple ion channels that have the capacity to generate action potentials in response to the extracellular glucose concentrations and ATP-sensitive potassium channels; these two pathways work in conjunction to stimulate the calcium hormone secretion. The regenerative model poses that at low levels glucose; the intracellular ATP/ADP are low, causing the ATP sensitive potassium channels to generate an action potentials, once this reaches -60mV, the voltage gated calcium channels (T-type ion channels) open; leading to the depolarisation of the membrane and activation of sodium and N-type voltage gated calcium channels this induction causes the secretion of glucagon.
The regenerative model, can be challenged with evidence from Rorsman and colleagues showing that and expression to suppressed concentration of glucose (6mM), alters the activity of the alpha secreting glucagon cells, reducing the frequency of action potentials, a reduction in P/Q-type calcium channels activity thereby inhibiting glucagon secretion. A second model suggests that exposure to high glucose concentration inhibits secretion of glucagon again via the ATP-sensitive potassium channel pathway.
Studies using human and mouse pancreatic alpha cells demonstrated that at low extracellular glucose concentrations, directly stimulates the secretion of glucagon and at high concentrations.
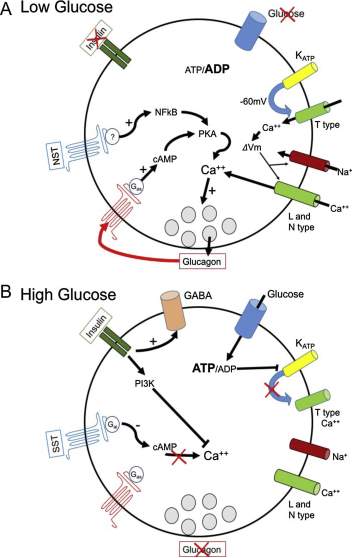
Figure 5: The regulation of glucagon secretion in low glucose concentration there is a decrease in ATP/ADP ration causing the ATP sensitive potassium channels to generate a membrane potential of -60mV causes depolarisation and opening of the T-type calcium ion channels, this causes an ‘domino affect’ resulting in the opening of sodium and high voltage I and N type calcium channels.
The rise in intracellular concentration of calcium causes (Synaptobrevin to no longer be bond to Synaptogamin, Synaptobrevin can now interact with T-snares on post synaptic membrane and glucagon is exocytose from the vesicles in alpha cell). The rise in intracellular calcium is also dependent on NTS secretion from delta cells, as well as glucagon itself (positive feedback loop- more glucagon secretion causes more stimulation for glucagon secretion). There is an inhibitory effect of insulin; there is a low secretion of insulin from the beta cells under low glucose concentrations.
At high glucose concentrations, the ATP/ ADP ration increases causes the closure of potassium ATP channels, resulting in the inactivation of calcium ion channels downstream, insulin is secreted by beta cells to reduced high glucose concentration in blood, but also has an inhibitor effect through P13K, causing a decreasing in calcium entry; as well as promoting the expression of GABA which inhibits glucagon secretion. Somatisation (secreted by Delta cells) active the Gαi pathway which will inhibit expression of the secondary messenger cAMP, thus preventing calcium influx.
How does glucagon affect the various systems of the body in the fed and fasted state?
Glucagon has many effects on several systems in the body, ranging from the cardiovascular, neurological, hepatic to the renal and intestinal.
In the cardiovascular system, pharmacological doses of glucagon activate Gαs pathway (noradrenaline binds to beta-adrenoreceptors) causing activation of funny current (If) which increases heart rate. This is because of an influx of calcium through VGCC’s; calcium will bind to ryanoid receptors in the SR, increasing the force of contraction in the heart; this effect is localised to the ventricles of the heart, because there are very few GCGR expression in the atrium. The effect of glucagon on the cardiovascular system , can further be analysed through the use of rat hearts; when perfused with levels of glucagon that induce physiological response (similar to the levels present in the human body) induced glycolysis and glucose metabolism, from this investigation it was identified the glucagon and insulin actions of the heart overlap in regards to fuel metabolism in the fed state (Ali and Drucker, 2009).
Glucagon stimulates AC and cAMP production in the nephrons, and in invitro experiments on human renal medullas (Yano et al., 2008). In the renal system; glucagon regulates GFR (glomerular filtration rate), urea excretion and water reabsorption in the descending LoH, these are all altered by the glucagon concentration both directly and indirectly. However, it has been shown that constant, long-term infusion of glucagon in mice leads to kidney damage through Hypertrophy resulting from hypertension (Li et al., 2008). The expression of glucagon in the gut is known motility, but very little is still known about the physiological role of glucagon in the gut (Ali and Drucker, 2009).
Glucagon and endocrine system
Glucagon is secreted by the α- cells in the islets; however, there is evidence to show that the glucagon receptor GCGR is present on beta, delta and cells’; this is why glucagon is thought to be co-regulated by insulin secretion (glucagon stimulates insulin secretion and vice versa) because its receptors are present on insulin secreting cells in the pancreas; studies have demonstrated that there is GCGR expression on rodent alpha cells and it is known that glucagon activates the G-alpha s pathway in alpha cell, thus increasing its own exocytosis suggesting that glucagon self-regulates its secretion from the pancreas (Röder et al., 2016).
Glucagon in the brain
The proglucagon gene is expressed in the brainstem, and to an extent in the hypothalamus (Abraham and Lam, 2016), as well as this, there are serval proglucagon-derived peptides in other regions of the brain (Ali and Drucker, 2009). Glucagon binds to its GCGR receptors on the brain membrane, stimulating AC produces cAMP. In rodents, pharmacological levels of glucagon in the brain have been shown to induce hyperglycaemia through stimulation of the cholinergic and adrenergic pathways. Furthermore, glucagon effects the CNS, and the HPG axis thus affecting the expression of the food hormone ghrelin (endogenous glucagon levels cause an increase in appetite in rats; high levels of glucagon cause the opposite – appetite suppression). These effects are of modulated by ghrelin and are classed a satiety-promoting effects of glucagon on the brain (Abraham and Lam, 2016).
Glucagon in adipose tissue
Glucagon receptors are present in the adipose tissue and induce lipolysis; this discovery is contradictory, as in humans and rat, and increase in glucagon infusion showed and increase in lipolysis in adipocytes; however, in human males, and increase in infusion of glucagon has no effect on lipolysis; hence the precise pathway of action of glucagon in the adipose tissue is still unknown (Ali and Drucker, 2009).
Glucagon in the hepatic system
A large majority of glucagon’s action occur in the liver. Once glucagon is secreted it enters the liver via the hepatic portal vein, it is first diluted by ‘glucagon-poor’ blood in circulation, this result n low glucagon levels (too low to induced lipolysis in adipose tissue); once the threshold of glucagon increases (usually twice or treble higher than its levels in other organs); it activate the Gαs pathway which activate PKA and induces glycogenolysis.
The purpose of glucagon (in the fasted state) is the medicate glucose production from glycerol (gluconeogenesis) and inhibit conversion of glucose to glycerol (glycogenesis) (Taborsky, 2010).
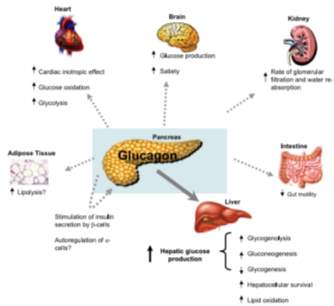
Figure 6: The effect of glucagon on the several organs: Brain, heart, kidney, intestines, adipose tissue and liver.
What is diabetes mellitus?
Diabetes mellitus as defined by WHO (world health organisation) is a metabolic disorder that is characterised by chronic hyperglycaemia with affects carbohydrate, fat and protein metabolism, long term effects include the dysfunction and failure of organs or organ systems. Diabetes mellitus can be broken down into several subsets; each referring to the aetiological genesis. The umbrella terms used when describing diabetes are Type 1, type 2 and Gestational diabetes mellitus.
Type 1 diabetes mellitus (T1DM) occurs if there is islet beta cell destruction, an autoimmune defect that causes destruction of the beta cell (leading to ketoacidosis); or failure of the beta-cell due to mitochondrial or genetic defects to sufficiently express the genes needed to synthesize and secrete insulin.
Type 2 diabetes mellitus (T2DM) is caused by a defect in insulin secretion (may also be causes by a resistance to insulin if secreted), or a defect in insulin receptors that prevent it from being recognized and thus does not decrease plasma glucose concentration. This causes either chronic hyperglycaemia or hypoglycaemia (if insulin is constantly being secreted even when plasma glucose is low).
Gestational diabetes mellitus is only seen in pregnant women and occurs as a result of a higher than normal glucose intake (to support the growing foetus) but an inability for this to be reduced even when insulin is secreted. For most gestational mothers; their fasting plasma glucose concentration returns to normal (> (5.6 mmol/L). If a woman has a fasting blood sugar of (5.6 to 6.9 mmol/L) and is considered pre-diabetic before pregnancy; during pregnancy it is likely she will develop gestational diabetes
Alpha cell in Type 1 and 2 diabetes mellitus
Alpha and beta cells in the islet of Langerhans share a common embryonic origin, giving beta cells the transdifferentiate ability to become alpha cells, and alpha cells have the same ability indicating the plasticity of pancreatic islet cells, this theory remains controversial.
Glucagon is secreted by the alpha cells and therefore is a principal hyperglycaemic hormone (Zhang et al., 2013), in diabetes glucagon secretion is not supressed (even at high plasma glucose concentration) this is known as glucagon secretion and causes hyperglycaemia. Conversely, at low levels of glucose, glucagon secretion is inadequate causing hypoglycaemia. These actions occur in conjunction with insulin therefore diabetes mellitus can be termed a bi-hormonal disorder involving both inadequate, defective or inappropriate insulin and glucagon secretion.
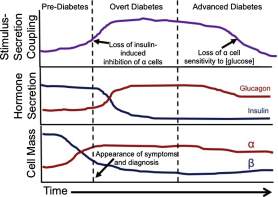
Figure 7: In the early stages of diabetes (pre-diabetes), there is gradual increase in inhibition of the alpha cell function; this increases exponentially well into overt diabetes, and upon entry into the stage of advance diabetes; there is a decline in loss of sensitivity to glucose by the alpha cell. Through the developmental stages of diabetes, there is a gradual increase of glucagon (this begins to plateau in the later stage of advanced diabetes; the reciprocal is true for insulin, there is a gradual decline in insulin secretion, well into the stages of advanced diabetes where the levels remain consistently low. The cell mass of alpha and beta cells parallels that of hormone secretion – there is a gradual increase in alpha cell mass expression (causes more glucagon to be secreted); whilst there is a decline in beta cell mass (resulting in the decline of insulin secretion) from overt to advanced diabetes mellitus.
Glucagon and its effects on Type 2 diabetes mellitus
There are several studies that have been conducted; that have identified some therapeutic uses of glucagon in the treatment of type 2 diabetes mellitus; one of which is the Role of KATP Channels in Glucose-Regulated Glucagon Secretion and Impaired Counter regulation in Type 2 Diabetes (Zhang et al., 2013); discovered that in controlling the potassium ATP channels, glucose regulation by glucagon secretion can be controlled in T2DM. the results showed that with the introduction of Tolbutamide ( a potassium channel blocker) led to a negative reciprocal expression of insulin and glucagon; insulin secretion increase when glucose was decreased from 6mM to 1mM; whilst glucagon secretion remained suppressed. Tolbutamide mimicked the inhibitory effects of glucose that is present in a non-diabetic, but fails to occur in a diabetic individual, whilst simultaneously, causing a 10 fold increase in insulin secretion preventing hyperglycaemia which in a type 2 diabatic can arise as a result of under-secretion of insulin to reduced plasma glucose concentration; or over secretion of glucagon irrespective of elevated plasma glucose concentration, tolbutamide reversed both of these actions. Although these results are of statistical significance (p
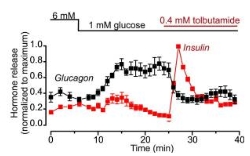
Figure 8: Graph showing the effect of a potassium channel agonist on glucagon and insulin secretion at 6Mm and 1mM glucose concentration.
NOTE: I hope to find another experimental paper (using human subjects with type two diabetes mellitus) and compare the results on the control of their insulin dependent diabetes when glucagon secretion is targeted.
Conclusion
Research has uncovered that diabetes mellitus (type I and II) are bi-hormonal diseases, that result from inappropriate insulin and glucagon secretions simultaneously. Targeting the mechanism of action in glucagon secretion can have some effect on the reduction in the severity of T2DM; however, these mechanisms that control glucagon (G-coupled pathway), therefore the pancreas would need to be targeted specifically and this is difficult.
Bibliography
Abraham, M.A., Lam, T.K.T., 2016. Glucagon action in the brain. Diabetologia 59, 1367–1371.
Ali, S., Drucker, D.J., 2009. Benefits and limitations of reducing glucagon action for the treatment of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 296, E415-421.
Ali, S., Lamont, B.J., Charron, M.J., Drucker, D.J., 2011. Dual elimination of the glucagon and GLP-1 receptors in mice reveals plasticity in the incretin axis. J. Clin. Invest. 121, 1917–1929.
Arifin, D.R., Bulte, J.W.M., 2011. Imaging of Pancreatic Islet Cells. Diabetes Metab. Res. Rev. 27, 761–766.
Authier, F., Desbuquois, B., 2008. Glucagon receptors. Cell. Mol. Life Sci. 65, 1880–1899.
Bonner, C., Kerr-Conte, J., Gmyr, V., Queniat, G., Moerman, E., Thévenet, J., Beaucamps, C., Delalleau, N., Popescu, I., Malaisse, W.J., Sener, A., Deprez, B., Abderrahmani, A., Staels, B., Pattou, F., 2015. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat. Med. 21, 512–517.
Bramswig, N.C., Everett, L.J., Schug, J., Dorrell, C., Liu, C., Luo, Y., Streeter, P.R., Naji, A., Grompe, M., Kaestner, K.H., 2013. Epigenomic plasticity enables human pancreatic α to β cell reprogramming. J. Clin. Invest. 123, 1275–1284.
El-Gohary1, Y., Gittes2, G., 2018. Structure of Islets and Vascular Relationship to the Exocrine Pancreas. Pancreapedia Exocrine Pancreas Knowl. Base.
Gallego, F.Q., Sinzato, Y.K., Miranda, C.A., Iessi, I.L., Dallaqua, B., Volpato, G.T., Scarano, W.R., SanMartín, S., Damasceno, D.C., 2018. Pancreatic islet response to diabetes during pregnancy in rats. Life Sci.
Richardson, C.C., Robinson, I.C.A.F., Low, M.J., Christie, M.R., Persaud, S.J., Jones, P.M., 2009. Somatostatin Secreted by Islet -Cells Fulfills Multiple Roles as a Paracrine Regulator of Islet Function. Diabetes 58, 403–411.
Hawkes, C.P., Lado, J.J., Givler, S., De Leon, D.D., 2018. The Effect of Continuous Intravenous Glucagon on Glucose Requirements in Infants with Congenital Hyperinsulinism. Springer Berlin Heidelberg, Berlin, Heidelberg.
Hellerström, C., 1983. The Biosynthesis of Glucagon, in: Lefèbvre, P.J. (Ed.), Glucagon I, Handbook of Experimental Pharmacology. Springer Berlin Heidelberg, Berlin, Heidelberg, pp. 121–138.
Insulin Synthesis and Secretion [WWW Document], n.d. URL http://www.vivo.colostate.edu/hbooks/pathphys/endocrine/pancreas/insulin.html (accessed 10.27.18).
Lamont, B.J., Li, Y., Kwan, E., Brown, T.J., Gaisano, H., Drucker, D.J., 2012. Pancreatic GLP-1 receptor activation is sufficient for incretin control of glucose metabolism in mice. J. Clin. Invest. 122, 388–402.
Li, X.C., Liao, T., Zhuo, J.L., 2008. Long-term hyperglucagonaemia induces early metabolic and renal phenotypes of Type 2 diabetes in mice. Clin. Sci. 114, 591–601.
Longuet, C., Robledo, A.M., Dean, E.D., Dai, C., Ali, S., McGuinness, I., de Chavez, V., Vuguin, P.M., Charron, M.J., Powers, A.C., Drucker, D.J., 2013. Liver-Specific Disruption of the Murine Glucagon Receptor Produces -Cell Hyperplasia: Evidence for a Circulating -Cell Growth Factor. Diabetes 62, 1196–1205.
Martin, B.C., Warram, J.H., al, et, 1992. Role of Glucose and Insulin Resistance in Development of Type 2 Diabetes Mellitus: Results of a 25-Year Follow-Up Study. Lancet Lond. 340, 925–9.
Mighiu, P.I., Yue, J.T.Y., Filippi, B.M., Abraham, M.A., Chari, M., Lam, C.K.L., Yang, C.S., Christian, N.R., Charron, M.J., Lam, T.K.T., 2013. Hypothalamic glucagon signaling inhibits hepatic glucose production. Nat. Med. 19, 766–772.
Miller, R.A., Shi, Y., Lu, W., Pirman, D.A., Jatkar, A., Blatnik, M., Wu, H., Cárdenas, C., Wan, M., Foskett, J.K., Park, J.O., Zhang, Y., Holland, W.L., Rabinowitz, J.D., Birnbaum, M.J., 2018. Targeting hepatic glutaminase activity to ameliorate hyperglycemia. Nat. Med. 24, 518–524.
Mokadem, M., Zechner, J.F., Margolskee, R.F., Drucker, D.J., Aguirre, V., 2014. Effects of Roux-en-Y gastric bypass on energy and glucose homeostasis are preserved in two mouse models of functional glucagon-like peptide-1 deficiency. Mol. Metab. 3, 191–201.
Mukharji, A., Drucker, D.J., Charron, M.J., Swoap, S.J., 2013. Oxyntomodulin increases intrinsic heart rate through the glucagon receptor. Physiol. Rep. 1.
Pancreas Location, Anatomy and Function in Digestion [WWW Document], n.d. URL https://healthjade.com/pancreas/ (accessed 10.27.18).
Pocai, A., Carrington, P.E., Adams, J.R., Wright, M., Eiermann, G., Zhu, L., Du, X., Petrov, A., Lassman, M.E., Jiang, G., Liu, F., Miller, C., Tota, L.M., Zhou, G., Zhang, X., Sountis, M.M., Santoprete, A., Capito’, E., Chicchi, G.G., Thornberry, N., Bianchi, E., Pessi, A., Marsh, D.J., SinhaRoy, R., 2009. Glucagon-Like Peptide 1/Glucagon Receptor Dual Agonism Reverses Obesity in Mice. Diabetes 58, 2258–2266.
Quiñones, M., Al-Massadi, O., Gallego, R., Fernø, J., Diéguez, C., López, M., Nogueiras, R., 2015. Hypothalamic CaMKKβ mediates glucagon anorectic effect and its diet-induced resistance. Mol. Metab. 4, 961–970.
Röder, P.V., Wu, B., Liu, Y., Han, W., 2016. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 48, e219.
Salem, V., Izzi-Engbeaya, C., Coello, C., Thomas, D.B., Chambers, E.S., Comninos, A.N., Buckley, A., Win, Z., Al-Nahhas, A., Rabiner, E.A., Gunn, R.N., Budge, H., Symonds, M.E., Bloom, S.R., Tan, T.M., Dhillo, W.S., 2016. Glucagon increases energy expenditure independently of brown adipose tissue activation in humans. Diabetes Obes. Metab. 18, 72–81.
Sipos, B., Sperveslage, J., Anlauf, M., Hoffmeister, M., Henopp, T., Buch, S., Hampe, J., Weber, A., Hammel, P., Couvelard, A., Höbling, W., Lieb, W., Boehm, B.O., Klöppel, G., 2015. Glucagon cell hyperplasia and neoplasia with and without glucagon receptor mutations. J. Clin. Endocrinol. Metab. 100, E783-788.
Sisley, S., Gutierrez-Aguilar, R., Scott, M., D’Alessio, D.A., Sandoval, D.A., Seeley, R.J., 2014. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J. Clin. Invest. 124, 2456–2463.
Svendsen, B., Larsen, O., Gabe, M.B.N., Christiansen, C.B., Rosenkilde, M.M., Drucker, D.J., Holst, J.J., 2018. Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell Rep. 25, 1127-1134.e2.
Taborsky, G.J., 2010. The Physiology of Glucagon. J. Diabetes Sci. Technol. 4, 1338–1344
THE ROLE OF GLUCAGON IN THE PATHOPHYSIOLOGY AND MANAGEMENT OF DIABETES [WWW Document], n.d. Clin. Case Rep. Endocr. Pract. URL /doi/abs/10.4158/EP15984.RA (accessed 10.27.18).
Thorel, F., Damond, N., Chera, S., Wiederkehr, A., Thorens, B., Meda, P., Wollheim, C.B., Herrera, P.L., 2011. Normal Glucagon Signaling and β-Cell Function After Near-Total α-Cell Ablation in Adult Mice. Diabetes 60, 2872–2882.
Type 2 diabetes [WWW Document], n.d. https://doi.org/10.1016/S0140-6736(17)30058-2
Ussher, J.R., Baggio, L.L., Campbell, J.E., Mulvihill, E.E., Kim, M., Kabir, M.G., Cao, X., Baranek, B.M., Stoffers, D.A., Seeley, R.J., Drucker, D.J., 2014. Inactivation of the cardiomyocyte glucagon-like peptide-1 receptor (GLP-1R) unmasks cardiomyocyte-independent GLP-1R-mediated cardioprotection. Mol. Metab. 3, 507–517.
Wu, Y., Li, J., Saleem, S., Yee, S.-P., Hardikar, A.A., Wang, R., 2010. c-Kit and stem cell factor regulate PANC-1 cell differentiation into insulin- and glucagon-producing cells. Lab. Investig. J. Tech. Methods Pathol. 90, 1373–1384.
Yano, Y., R Cesar, K., Araujo, M., Rodrigues, A., Andrade, L., Magaldi, A., 2008. Aquaporin 2 expression increased by glucagon in normal rat inner medullary collecting ducts. Am. J. Physiol. Renal Physiol. 296, F54-9.
Yi, F., Sun, J., Lim, G.E., Fantus, I.G., Brubaker, P.L., Jin, T., 2008. Cross Talk between the Insulin and Wnt Signaling Pathways: Evidence from Intestinal Endocrine L Cells. Endocrinology 149, 2341–2351.
Yosten, G.L.C., 2018. Alpha cell dysfunction in type 1 diabetes. Peptides, Newer peptide-based agents for treatment of patients with Type 2 diabetes 100, 54–60.
Zhang, Q., Ramracheya, R., Lahmann, C., Tarasov, A., Bengtsson, M., Braha, O., Braun, M., Brereton, M., Collins, S., Galvanovskis, J., Gonzalez, A., Groschner, L.N., Rorsman, N.J.G., Salehi, A., Travers, M.E., Walker, J.N., Gloyn, A.L., Gribble, F., Johnson, P.R.V., Reimann, F., Ashcroft, F.M., Rorsman, P., 2013. Role of KATP Channels in Glucose-Regulated Glucagon Secretion and Impaired Counterregulation in Type 2 Diabetes. Cell Metab. 18, 871–882.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Diabetes"
Diabetes is a metabolic disorder that results in an abnormally high blood glucose level. Blood glucose levels are controlled by insulin produced by the pancreas. In diabetics, the pancreas either doesn’t produce enough (or any) insulin, or the body does not respond sufficiently to the insulin that the pancreas produces.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: