Posttranslational Modifications of Histone Proteins
Info: 7148 words (29 pages) Dissertation
Published: 21st Feb 2022
1. Introduction
The elucidation of the double helical DNA structure stands as one of the biggest discoveries in modern science. This seminal discovery along with findings of Avery1 and Hershey-Chase2 confirmed the role of DNA as the genetic material of life. Based on this and other scientific findings of the time, Crick introduced the idea of the “central dogma of molecular biology” to explain the flow of information in biological systems.3 According to the central dogma, genetic information is maintained through semi-conservative replication of DNA and is sequentially propagated by two templated processes, the transcription of DNA to RNA, followed by translation of RNA to produce proteins. The dogma allows the possibility of feedback from the RNA to the DNA level, but discards such a transfer from protein to DNA or protein to protein. In some way, it emphasizes most of the genetic regulation to occur at the DNA level, which holds true to explain many biological situations.
However, it is not always possible to explain the genetic complexity observed in biological systems using the mere genetic code of DNA, encoded by 4 letters (A, T, G, C). One of the simplest and straight-forward example to illustrate this complexity is the phenomenon of cell differentiation and development. As one moves from simple unicellular organisms to multicellular organisms, like human, one observes that despite identical genome sequence, the cells differentiate into over 200 distinct cells types. The DNA sequence alone cannot explain this phenomenon, since all cells contain nearly identical genomic information, and suggests a regulation of gene expression without alterations in the DNA sequence. The discovery of prions4 and certain non-Mendelian inheritance patterns further corroborated the idea of an additional layer of regulation above the genetic level (‘Epigenetics’).
Indeed, there exist many regulatory mechanisms which provide multidimensional layers of regulation for genomic sequences without physically changing them. This epigenetic regulation occurs through modulation of the chromatin template, the physiological form of genomic DNA.
1.1 The chromatin template
In eukaryotes, the chromosomal DNA is packaged in a dynamic polymeric complex called chromatin. The fundamental unit of chromatin is the nucleosome, composed of an octameric protein core around which ~ 150 base pairs of DNA is spooled in a left-handed spiral (Figure 1). This wrapping of DNA around histone octamer constitutes the first level of genomic compaction. Although the composition of the nucleosome was known for a while, the first insight into the nucleosome structure was provided via 2.8 Å crystal structure solved by Luger et al.5in 1997. The octameric protein core is made up of two copies of four relatively small (10-20 kDa), highly basic histone proteins – H2A, H2B, H3, and H4. The canonical histone proteins contain a histone-fold motif, made up of three α helices with one central helix connected to two other small helices through two loops, along with flexible N-terminal tails. Histone protein, H2A has an additional C-terminal tail. The histone-fold motifs of distinct histone proteins complement each other to form a handshake motif leading to assembly of histone heterodimers (H2A/H2B and H3/H4). For octamer constitution, two H3/H4 heterodimers come together to form a central (H3/H4)2 tetramer with two binding surfaces for H2A/H2B heterodimers on opposite faces. The DNA is wrapped around the octamer in a bent duplex conformation, an energetically unfavorable conformation owing to phosphate backbone interactions of DNA.6 However, this energetically unfavorable situation is compensated by highly favorable histone-DNA interactions of the nucleosome leading to very efficient packaging of the DNA. These favorable interactions between histone and DNA stem from ionic interactions, water-mediated hydrogen bonds, non-ionic interactions, and dipole interaction between histone-fold helices and phosphate backbone.7,8
The next level of genomic compaction occurs through self-assembly of nucleosomes into higher-order chromatin structures. Initially, individual nucleosome core particles (NCPs) thread together with adjacent nucleosome core particles through a linker DNA to form the 11-nm ‘bead on a string’ template.9 The 11-nm chromatin template is further compacted into a 30-nm higher-order structure through recruitment of an additional histone protein, H1.10 The H1 protein promotes genomic compaction by interactions between its central globular domain (gH1) and nucleosome core particles. There are currently two different models explaining the interaction between gH1 and NCP. First, the symmetric model, which assumes gH1 to bind at the NCP dyad and interact with linker DNA extending from both side of NCP.11,12 The second model considers gH1 to bind near the dyad and interact with 10-20 bp of linker DNA extending from one side of NCP.11,13,14
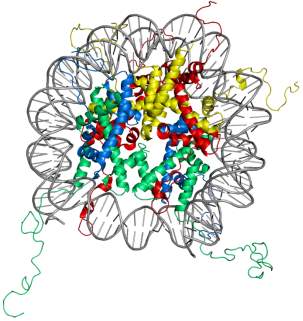
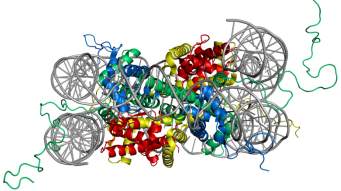
Figure 1. Structure of the nucleosome core particle. The figure on the right shows the top view and on the left shows the side view of nucleosome structure. The histone proteins H2A, H2B, H3, and H4 are shown in red, yellow, green and blue whereas DNA is shown in grey. Adapted from PDB structure: 1KX5.15
Despite recent studies having questioned the prevalence of the 30-nm chromatin fiber, structural details regarding the organization of the 30 nm fiber have been one of the most debated topics in chromatin biology.16,17 Two models have been suggested – the solenoid (one-start) model, wherein sequential nucleosomes are coiled around a helical axis, or the zig-zag (two-start) model, wherein sequential nucleosomes are separated by a zig-zag pattern of straight linker DNA.18,19 A recent cryo-EM structure of 30 nm fibers shows a left-handed parallel double helix supporting the two-start model proposed by Richmond and colleagues.20 It is worth-noting that the DNA is compacted only approximately 50-fold by 30 nm fiber and higher-order re-organization exists for further compaction which are yet to be resolved. The efficiency of DNA packaging in nucleosome can be gauged by the fact that human genomic DNA which spans over a length of two meters is compacted 10000-20000 fold to fit in the cell nucleus.21
1.2 Epigenetic mechanisms
The tight compaction of the eukaryotic genomic DNA into chromatin leads to a condensed DNA structure which occludes DNA-binding proteins. However, a rigid chromatin structure can only block DNA-templated processes rather than regulate it. Indeed, chromatin is dynamic in nature and allows DNA-binding proteins selective access to the nucleosomal DNA via different epigenetic mechanisms. These comprise mainly of: 1) Modification of DNA bases,22,23 2) Post-translational modification of histone proteins,24 3) Introduction of histone variants,25-28 4) Nucleosome positioning,29-32 5) Non-coding RNA based mechanism.33-35 In this thesis, we focus on the role of the post-translational modification of histone protein in epigenetics.
1.3 Post-translational modifications of histone proteins
Histone proteins were first found to be acetylated by Allfrey in 1964.36 Since then a large number of histone protein modifications have been identified using western blotting and mass spectrometry based methods.37 These include lysine acetylation, acylation with longer chain length acids,38,39 formylation,40 methylation,41,42 sumoylation, and ubiquitination;43 arginine methylation and citrullination;44 serine and threonine phosphorylation and GlcNAcylation;45-47 histidine phosphorylation;48 glutamine methylation;49 glutamate and arginine mono- and poly-ADP ribosylation,24 and tyrosine hydroxylation. The chemical structure of few important and frequently studied histone modifications are shown in Figure 2.
Histone PTMs are highly dynamic in nature and are regulated by the opposing catalytic activity of enzymes. Histone proteins are modified by transferase ‘writers’ whereas the reverse reaction is catalysed by a variety of ‘eraser’ enzymes. For example, in the case of histone acetylation, histone acetyltransferases (HATs) ‘writers’ catalyse acetylation of lysine residues whereas histone deacetylases (HDACs) erase this modification by hydrolysis. It is striking to note the diverse chemical space of the post-translational modifications. PTMs size varies vastly from small a methyl group to the addition of a whole protein via ubiquitination/sumoylation, they can modulate electrostatics properties of proteins by removing or adding charges as in acetylation or phosphorylation, or they can add hydrogen bonding donors/acceptors such as O-GlcNAc. Indeed, these diverse modifications play an important and context-dependent role in epigenetic regulation.
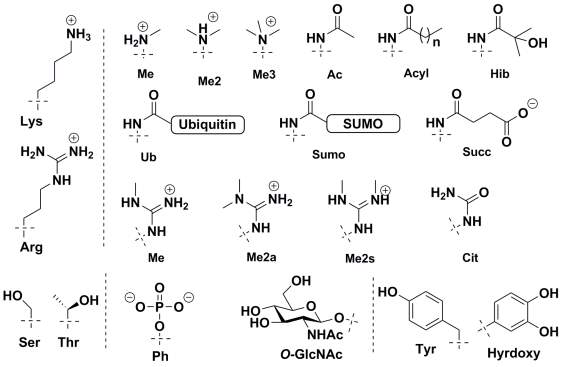
Figure 2. Chemical Structure of commonly observed histone post-translational modifications.
1.3 Role of Histone PTMs in epigenetics
The epigenetic regulation by histone protein modifications, in their own capacity, occurs through two main mechanisms.50,51 The first mechanism involves modulation of nucleosome structure/stability upon histone modification.52 This structure modulation can stem from the changes in histone-fold or histone-DNA interactions. The second and widely prevalent mechanism involves the recruitment of specific reader or effector proteins to the site of modification which either catalyze the functional output of the modification or recruit additional chromatin remodelers.53,54 Interestingly, different histone protein modifications can also enhance or interfere with their respective signal output (histone cross-talk) to provide an additional layer of complexity to transcriptional regulation.
1.3.1 Nucleosome structural modulation
Histone PTMs can directly modulate the nucleosome structure to a subtle or drastic extent either through electrostatic interactions or via sterics depending on the modification and site (Figure 3). For long, histone acetylation has been associated with a relaxed chromatin state, facilitating transcription, although the extent of this ‘relaxation’ has just started to be understood. In a simple mechanism, acetylation of lysine residue inhibits the salt-bridge interaction between positively-charged lysine and negatively-charged nucleosomal DNA leading to a less tightly packed chromatin. A more open chromatin environment provides RNA polymerases easy access to the DNA template for transcription. Indeed, in line with this simple mechanism, histone acetylation marks are found to be enriched at enhancer and promoter regions.55,56 In a recent study, details at the molecular level linking H3K56 acetylation to chromatin accessibility were provided. Separate experiments based on mutational analysis and synthetic nucleosome construct bearing homogeneous acetylation revealed that although acetylation does not affect the nucleosome structure substantially, it does promotes DNA unwrapping leading to nucleosome-free chromatin regions.57-59 Consistent with this a fluorescence resonance energy transfer (FRET)-based study revealed destabilization of nucleosome structure upon H3K64 acetylation leading to enrichment of the mark at active genes. The destabilization was attributed to reduced histone-DNA interaction.60 In a slightly different mechanism, acetylation of H4-K91 was shown to destabilize the nucleosome structure by modulating the H2A/B dimer-H3/H4 tetramer interface. This is of special interest since here it’s the steric of the modification rather than electrostatic effect, usually associated with acetylation, which is responsible for the functional output.61
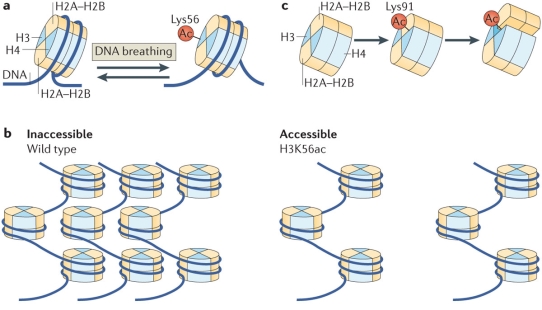
Figure 3. Regulation of nucleosome structure by histone PTMs through histone-histone and histone-DNA interactions. (a) H3K56-acetylation enhances DNA unwrapping by regulating histone-DNA interactions. (b) H3K56-acetylation promotes nucleosome-free higher order chromatin structure. (c)H4K91-acetylation destabilizes the nucleosome stability. (Reproduced from Tessarz et al.52)
There lies a potential that negative charge introduced on histone protein through modifications such as phosphorylation and ADP-ribosylation can affect histone-DNA interactions mediated by electrostatic repulsion, and thus regulate nucleosome structure. Indeed, phosphorylation of H3T118 site, located in vicinity of nucleosomal dyad (within 3 Å), destabilizes the nucleosome structure.62 In a study based on in vitro nucleosome reconstitutions, North et al. reporteddramatically reduced DNA-histone binding free energy and decreased nucleosome stability upon phosphorylation of H3T118. In line with destabilized nucleosome structure, a significant increase in DNA accessibility near the nucleosome dyad was found. It is worth-noting that the H3(T118E) substitution, a mutation widely used to mimic phosphates, did not affect the DNA-histone binding free energy or nucleosome mobility. Interestingly, a separate report from the same group reported formation of an alternative nucleosome structure upon H3T118 phosphorylation highlighting significant structure distortion.63
In contrast to these histone PTMs which modulate the charge of amino acids, there are neutral modifications which can modulate nucleosome structure based on their steric properties. Certainly modifications such as ubiquitination and sumoylation can affect histone-histone or histone-DNA leading to conformational change of the nucleosome. In a previous report, it has been speculated that ubiquitin modification of H4K91 likely alters the nucleosome structure by destabilizing the histone dimer-tetramer interface.64
Intriguingly, contrary to earlier belief that histone methylation cannot directly perturb nucleosome structure,50 recent results have shown otherwise. Asymmetric dimethylation of H3R42 residue supposedly affects the histone-DNA interaction by blocking hydrogen bonding. The reduced histone-DNA interaction lowers the overall nucleosome stability and promotes transcription.65 Similarly, for H3K79 methylation, a subtle reorganization around the modification occurs because of a loss of a hydrogen bond to histone protein, H4.66
1.3.2 Recruitment of effector proteins
The second mechanism by which histone PTMs exert their function is by acting as a docking site for numerous chromatin-associated protein complexes.50,53 These nucleosome-binding complexes contain distinct domains which can recognize specific histone modifications. Often, these protein complexes are multivalent and have more than one domain through which they can recognize several distinct modifications.67 Once localized to a genomic region by the PTM, these chromatin-associated proteins can recruit other nuclear signaling machinery to catalyze DNA-templated processes.
Of all the histone modifications, protein domains recognizing lysine methylation are best understood and include PHD (plant homeodomain) fingers, ADD (ATRX-DNMT3-DNMT3L), BAH (bromo-adjacent homology), chromodomain and Tudor domains.68-70 Lysine methylation marks can activate or repress transcription, depending on methylation state and site of modification. Interestingly, different domains recognizing the same modification can be found in transcription activating or repressing complexes, which suggests a context-dependent readout for the PTMs.71 For example, H3K4me3, considered to be a gene-activation mark, binds to TFIID through its PHD finger, implicating it in transcriptional activation.72 However, recognition of the same mark by PHD finger of ING2 links the modification to gene repression.73 In separate studies, the same modification has also been shown to bind to the tandem chromodomains within CHD1, a chromatin remodeler and to the tandem Tudor domains within JMJD2A, a histone demethylase.74,75 The recruitment of effector proteins to other methylation marks has been linked to chromatin compaction. In a relevant example, binding of HP1 protein through its chormodomain to H3K9me3 modification, a repressive mark, has been found to affect the heterochromatin structure.76,77
Histone acetylation can be recognized by three different reader domains: the bromodomain78,79, the double PHD finger (DPF) of DPF3b protein and the double pleckstrin homology (PH) domain of the histone chaperone Rtt106 can also specifically recognize acetylated histones.80-82 Similarly, there are reader domains such as 14-3-3 and BRCT for phosphorylated histone proteins.83,84
1.3.3 Histone Cross-talk
In a biologically-relevant context, nucleosomes are decorated with different histone modifications at multiple sites, rather than bearing a single histone modification.85 Given the number of possible histone modifications and modification sites available on nucleosome, the number of potential combinations is astronomical. These modification combinations can potentially act as a mechanism for tighter control of chromatin regulation, where a ‘codon’ of modifications can translate a certain epigenetic state. Different modifications can work in a cohort and interact with each other (‘cross-talk’) to provide an extra-layer of genomic regulation.86
There are various mechanisms possible for this histone cross-talk.50,51,87-89 In one of the simplest mechanisms, two or more different histone modifications can compete for the same modification site leading to a competitive antagonism to define the epigenetic state. This kind of cross-talk can operate for histone residues which are capable of different modifications such as lysine which can undergo different modifications such as acetylation, methylation (mono-, di-, and tri-), ubiquitination, various acylations, and sumoylation in a context-dependent manner.
In a different mechanism, the state of modification at one site can promote or inhibit modification at other site. Previous studies have shown that ubiquitination of H2BK123 by Rad6 enzyme is a necessary condition for H3K4 methylation catalyzed by the COMPASS sub-complex and H3K79 methylation mediated by the Dot1 methyltransferase.90,91 In a similar fashion, H3S10 phosphorylation mediated by the Snf1 kinase is known to promote H3K14 acetylation by the Gcn5 acetyltransferase, enhancing gene activation.92 Daujat et al. have previously shown that acetylation at H3K18 and H3K23 induces the methylation of H3R17 residue by the CARM1 methyltransferase, resulting in activation of estrogen responsive genes.93 Note that in the last two examples, histone modification of one site promote other modification on the same histone protein (cis-histone effect), whereas in the first example, the modification of one histone proteins affects modification of a different histone protein (trans-histone effect). However, the modifications can also be negatively cooperative. In one of the classical examples of histone cross-talk, H3S10 phosphorylation inhibits binding of HP1 protein to di- and tri-methylated H3K9 during mitosis.94 Similarly, H3K4 acetylation inhibits binding of Chp1 protein to di- and tri-methylated H3K9 in yeast to regulate heterochromatin assembly.95
Alternatively, different histone modifications can jointly amplify the binding efficiency of effector proteins. Using MS-based quantitative interaction proteomics, Vermeulen et al. showed that the binding of PHF8 to H3K4me3 is stronger in presence of acetylated H3K9 and H3K14.96 Apart from cross-talk between histone proteins, there can be a cross-talk between modifications of histone protein and nucleosomal DNA. In an earlier report from the Kouzarides group, the binding of UHRF1 protein to methylated nucleosome was shown to be significantly enhanced upon CpG methylation of nucleosomal DNA.97
1.4 Goals of the work of this thesis
The overall goal of my doctoral work was to use the tools of chemical biology to decipher how different posttranslational modifications of histone proteins function in epigenetic regulation. Special emphasis was given to understand the role of O-GlcNAcylation in transcriptional regulation; since not much is known about the functional consequence of this recently discovered histone modification. In the past, most chromatin research to understand the role of histone PTMs has relied on the use of synthetic histone peptides. However, these peptides are mere fragments of the physiologically-relevant substrate, the nucleosome, and cannot be used to investigate epigenetic processes which rely on the multivalency arising from histone-histone and histone-DNA interactions.98,99
In this thesis, we applied protein chemical reactions to synthesize homogeneously modified histone proteins, which were then used to study the effect of PTMs on nucleosome biophysics and the nucleosome interactome. For the study of histone O-GlcNAcylation, three different modification sites were chosen (Chapter 2, 3, 4). All three sites were situated on distinct nucleosome surfaces, raising the possibility of context-dependent functional outcome. The effect of the modifications on the nucleosome structure was probed using different biophysical methods, including circular dichroism spectroscopy and native mass spectrometry.
Recent advances in quantitative mass spectrometry has made it the method of choice for studying the ‘reader/effector’ proteins for modified nucleosome, over traditional western-blotting based methods.37,100 We applied MS-based affinity proteomics to identify putative interaction partners for modified nucleosomes based on which we could propose a molecular mechanism for the functional output.
In the last chapter, we envisioned to study the cross-talk between O-GlcNAcylation and phosphorylation. Use of dehydroalanine as a chemical handle to readily install different chemical modifications proved effective, and could potentially be used for study of any such cross-talk.
1.5 References
1 Avery, O. T., MacLeod, C. M. & McCarty, M. STUDIES ON THE CHEMICAL NATURE OF THE SUBSTANCE INDUCING TRANSFORMATION OF PNEUMOCOCCAL TYPES : INDUCTION OF TRANSFORMATION BY A DESOXYRIBONUCLEIC ACID FRACTION ISOLATED FROM PNEUMOCOCCUS TYPE III. The Journal of Experimental Medicine 79, 137-158, (1944).
2 Hershey, A. D. & Chase, M. INDEPENDENT FUNCTIONS OF VIRAL PROTEIN AND NUCLEIC ACID IN GROWTH OF BACTERIOPHAGE. The Journal of General Physiology 36, 39-56, (1952).
3 Crick, F. Central Dogma of Molecular Biology. Nature 227, 561-563, (1970).
4 Shorter, J. & Lindquist, S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet 6, 435-450, (2005).
5 Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature (London) 389, 251-260, (1997).
6 Richmond, T. J. & Davey, C. A. The structure of DNA in the nucleosome core. Nature (London, U. K.) 423, 145-150, (2003).
7 McGinty, R. K. & Tan, S. Nucleosome Structure and Function. Chemical Reviews 115, 2255-2273, (2015).
8 Davey, C. A., Sargent, D. F., Luger, K., Maeder, A. W. & Richmond, T. J. Solvent Mediated Interactions in the Structure of the Nucleosome Core Particle at 1.9 Resolution. J. Mol. Biol. 319, 1097-1113, (2002).
9 Olins, A. L. & Olins, D. E. Spheroid chromatin units (v bodies). Science (New York, N.Y.) 183, 330-332, (1974).
10 Harshman, S. W., Young, N. L., Parthun, M. R. & Freitas, M. A. H1 histones: current perspectives and challenges. Nucleic Acids Res. 41, 9593-9609, (2013).
11 Fan, L. & Roberts, V. A. Complex of linker histone H5 with the nucleosome and its implications for chromatin packing. Proc. Natl. Acad. Sci. U. S. A. 103, 8384-8389, (2006).
12 Syed, S. H. et al. Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc. Natl. Acad. Sci. U. S. A. 107, 9620-9625, S9620/9621-S9620/9610, (2010).
13 Zhou, B.-R. et al. Histone H4 K16Q Mutation, an Acetylation Mimic, Causes Structural Disorder of Its N-Terminal Basic Patch in the Nucleosome. J. Mol. Biol. 421, 30-37, (2012).
14 Zhou, Y. B., Gerchman, S. E., Ramakrishnan, V., Travers, A. & Muyldermans, S. Position and orientation of the globular domain of linker histone H5 on the nucleosome. Nature (London) 395, 402-405, (1998).
15 Davey, C. A., Sargent, D. F., Luger, K., Maeder, A. W. & Richmond, T. J. Solvent Mediated Interactions in the Structure of the Nucleosome Core Particle at 1.9 Å Resolution. Journal of Molecular Biology 319, 1097-1113, (2002).
16 Eltsov, M., MacLellan, K. M., Maeshima, K., Frangakis, A. S. & Dubochet, J. Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc. Natl. Acad. Sci. U. S. A. 105, 19732-19737, (2008).
17 Maeshima, K., Hihara, S. & Eltsov, M. Chromatin structure: does the 30-nm fiber exist in vivo? Curr. Opin. Cell Biol. 22, 291-297, (2010).
18 Robinson, P. J. J., Fairall, L., Huynh, V. A. T. & Rhodes, D. EM measurements define the dimensions of the “30-nm” chromatin fiber: evidence for a compact, interdigitated structure. Proc. Natl. Acad. Sci. U. S. A. 103, 6506-6511, (2006).
19 Schalch, T., Duda, S., Sargent, D. F. & Richmond, T. J. X-ray structure of a tetranucleosome and its implications for the chromatin fiber. Nature (London, U. K.) 436, 138-141, (2005).
20 Song, F. et al. Cryo-EM Study of the Chromatin Fiber Reveals a Double Helix Twisted by Tetranucleosomal Units. Science (New York, N.Y.) 344, 376-380, (2014).
21 Woodcock, C. L. & Ghosh, R. P. Chromatin Higher-order Structure and Dynamics. Cold Spring Harbor Perspectives in Biology 2, (2010).
22 Song, C. X., Yi, C. & He, C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nature biotechnology 30, 1107-1116, (2012).
23 Klose, R. J. & Bird, A. P. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 31, 89-97, (2006).
24 Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381-395, (2011).
25 Talbert, P. B. & Henikoff, S. Histone variants – ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 11, 264-275, (2010).
26 Weber, C. M. & Henikoff, S. Histone variants: dynamic punctuation in transcription. Genes Dev. 28, 672-682, (2014).
27 Fan, J. Y., Gordon, F., Luger, K., Hansen, J. C. & Tremethick, D. J. The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states. Nat. Struct. Biol. 9, 172-176, (2002).
28 Chakravarthy, S., Patel, A. & Bowman, G. D. The basic linker of macroH2A stabilizes DNA at the entry/exit site of the nucleosome. Nucleic Acids Res. 40, 8285-8295, (2012).
29 Jiang, C. & Pugh, B. F. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10, 161-172, (2009).
30 Clapier, C. R. & Cairns, B. R. The biology of chromatin remodeling complexes. Annu Rev Biochem 78, 273-304, (2009).
31 Ho, L. & Crabtree, G. R. Chromatin remodelling during development. Nature 463, 474-484, (2010).
32 Narlikar, G. J., Sundaramoorthy, R. & Owen-Hughes, T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154, 490-503, (2013).
33 Lee, J. T. Epigenetic regulation by long noncoding RNAs. Science (New York, N.Y.) 338, 1435-1439, (2012).
34 Bhan, A. & Mandal, S. S. Long Noncoding RNAs: Emerging Stars in Gene Regulation, Epigenetics and Human Disease. ChemMedChem 9, 1932-1956, (2014).
35 Mercer, T. R. & Mattick, J. S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol 20, 300-307, (2013).
36 Allfrey, V. G., Faulkner, R. & Mirsky, A. E. ACETYLATION AND METHYLATION OF HISTONES AND THEIR POSSIBLE ROLE IN THE REGULATION OF RNA SYNTHESIS. Proc Natl Acad Sci U S A 51, 786-794, (1964).
37 Huang, H., Lin, S., Garcia, B. A. & Zhao, Y. Quantitative Proteomic Analysis of Histone Modifications. Chemical Reviews 115, 2376-2418, (2015).
38 Tan, M. et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016-1028, (2011).
39 Dai, L. et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat Chem Biol 10, 365-370, (2014).
40 Jiang, T., Zhou, X., Taghizadeh, K., Dong, M. & Dedon, P. C. N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc Natl Acad Sci U S A 104, 60-65, (2007).
41 Martin, C. & Zhang, Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6, 838-849, (2005).
42 Black, J. C., Van Rechem, C. & Whetstine, J. R. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 48, 491-507, (2012).
43 Weake, V. M. & Workman, J. L. Histone ubiquitination: triggering gene activity. Mol Cell 29, 653-663, (2008).
44 Christophorou, M. A. et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 507, 104-108, (2014).
45 Fong, J. J. et al. β-N-Acetylglucosamine (O-GlcNAc) Is a Novel Regulator of Mitosis-specific Phosphorylations on Histone H3. Journal of Biological Chemistry 287, 12195-12203, (2012).
46 Sakabe, K., Wang, Z. & Hart, G. W. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci U S A 107, 19915-19920, (2010).
47 Fujiki, R. et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 480, 557-560, (2011).
48 Kee, J.-M., Villani, B., Carpenter, L. R. & Muir, T. W. Development of Stable Phosphohistidine Analogues. Journal of the American Chemical Society 132, 14327-14329, (2010).
49 Tessarz, P. et al. Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature 505, 564-568, (2014).
50 Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res 21, 381-395, (2011).
51 Kouzarides, T. Chromatin Modifications and Their Function. Cell 128, 693-705, (2007).
52 Tessarz, P. & Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 15, 703-708, (2014).
53 Musselman, C. A., Lalonde, M.-E., Cote, J. & Kutateladze, T. G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218-1227, (2012).
54 Taverna, S. D., Li, H., Ruthenburg, A. J., Allis, C. D. & Patel, D. J. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025-1040, (2007).
55 Kuo, M. H. & Allis, C. D. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays : news and reviews in molecular, cellular and developmental biology 20, 615-626, (1998).
56 Wang, Z. et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature genetics 40, 897-903, (2008).
57 Neumann, H. et al. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell 36, 153-163, (2009).
58 Simon, M. et al. Histone fold modifications control nucleosome unwrapping and disassembly. Proceedings of the National Academy of Sciences 108, 12711-12716, (2011).
59 Watanabe, S. et al. Structural characterization of H3K56Q nucleosomes and nucleosomal arrays. Biochim. Biophys. Acta, Gene Regul. Mech. 1799, 480-486, (2010).
60 Di Cerbo, V. et al. Acetylation of histone H3 at lysine 64 regulates nucleosome dynamics and facilitates transcription. eLife 3, e01632, (2014).
61 Ye, J. et al. Histone H4 lysine 91 acetylation a core domain modification associated with chromatin assembly. Mol Cell 18, 123-130, (2005).
62 North, J. A. et al. Phosphorylation of histone H3(T118) alters nucleosome dynamics and remodeling. Nucleic Acids Research 39, 6465-6474, (2011).
63 North, J. A. et al. Histone H3 phosphorylation near the nucleosome dyad alters chromatin structure. Nucleic Acids Research 42, 4922-4933, (2014).
64 Yan, Q. et al. BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol Cell 36, 110-120, (2009).
65 Casadio, F. et al. H3R42me2a is a histone modification with positive transcriptional effects. Proceedings of the National Academy of Sciences 110, 14894-14899, (2013).
66 Lu, X. et al. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat Struct Mol Biol 15, 1122-1124, (2008).
67 Fierz, B. & Muir, T. W. Chromatin as an expansive canvas for chemical biology. Nat Chem Biol 8, 417-427, (2012).
68 Champagne, K. S. & Kutateladze, T. G. Structural insight into histone recognition by the ING PHD fingers. Current drug targets 10, 432-441, (2009).
69 Kim, J. et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO reports 7, 397-403, (2006).
70 Maurer-Stroh, S. et al. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends in Biochemical Sciences 28, 69-74, (2003).
71 Wu, J. I., Lessard, J. & Crabtree, G. R. Understanding the words of chromatin regulation. Cell 136, 200-206, (2009).
72 Vermeulen, M. et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58-69, (2007).
73 Shi, X. et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96-99, (2006).
74 Huang, Y., Fang, J., Bedford, M. T., Zhang, Y. & Xu, R.-M. Recognition of Histone H3 Lysine-4 Methylation by the Double Tudor Domain of JMJD2A. Science (New York, N.Y.) 312, 748-751, (2006).
75 Sims, R. J. et al. Human but Not Yeast CHD1 Binds Directly and Selectively to Histone H3 Methylated at Lysine 4 via Its Tandem Chromodomains. Journal of Biological Chemistry 280, 41789-41792, (2005).
76 Bannister, A. J. et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120-124, (2001).
77 Lachner, M., O’Carroll, D., Rea, S., Mechtler, K. & Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116-120, (2001).
78 Dhalluin, C. et al. Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491-496, (1999).
79 Sanchez, R. & Zhou, M.-M. The role of human bromodomains in chromatin biology and gene transcription. Current opinion in drug discovery & development 12, 659-665, (2009).
80 Lange, M. et al. Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes & Development 22, 2370-2384, (2008).
81 Zeng, L. et al. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature 466, 258-262, (2010).
82 Su, D. et al. Structural basis for recognition of H3K56-acetylated histone H3-H4 by the chaperone Rtt106. Nature 483, 104-107, (2012).
83 Macdonald, N. et al. Molecular basis for the recognition of phosphorylated and phosphoacetylated histone h3 by 14-3-3. Mol Cell 20, 199-211, (2005).
84 Stucki, M. et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213-1226, (2005).
85 Shema, E. et al. Single-molecule decoding of combinatorially modified nucleosomes. Science (New York, N.Y.) 352, 717-721, (2016).
86 Berger, S. L. The complex language of chromatin regulation during transcription. Nature 447, 407-412, (2007).
87 Lee, J.-S., Smith, E. & Shilatifard, A. The Language of Histone Crosstalk. Cell 142, 682-685.
88 Suganuma, T. & Workman, J. L. Crosstalk among Histone Modifications. Cell 135, 604-607, (2008).
89 Suganuma, T. & Workman, J. L. Signals and Combinatorial Functions of Histone Modifications. Annual Review of Biochemistry 80, 473-499, (2011).
90 Kim, J. et al. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell 137, 459-471, (2009).
91 Lee, J. S. et al. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell 131, 1084-1096, (2007).
92 Walter, W. et al. 14-3-3 Interaction with Histone H3 Involves a Dual Modification Pattern of Phosphoacetylation. Molecular and Cellular Biology 28, 2840-2849, (2008).
93 Daujat, S. et al. H3K64 trimethylation marks heterochromatin and is dynamically remodeled during developmental reprogramming. Nat. Struct. Mol. Biol. 16, 777-781, (2009).
94 Fischle, W. et al. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116-1122, (2005).
95 Xhemalce, B. & Kouzarides, T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes & Development 24, 647-652, (2010).
96 Vermeulen, M. et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142, 967-980, (2010).
97 Bartke, T. et al. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143, 470-484, (2010).
98 Allis, C. D. & Muir, T. W. Spreading Chromatin into Chemical Biology. ChemBioChem 12, 264-279, (2011).
99 Müller, M. M. & Muir, T. W. Histones: At the Crossroads of Peptide and Protein Chemistry. Chemical Reviews 115, 2296-2349, (2015).
100 Eberl, H. C., Mann, M. & Vermeulen, M. Quantitative Proteomics for Epigenetics. ChemBioChem 12, 224-234, (2011).
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Chemistry"
Chemistry is a science involving the study of the elements and matter at the atomic and molecular level including their composition, structure, properties, behaviour, and how they react or combine.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: