Growth, Spectral, Nonlinear and DFT Computations of a Third Order Nonlinear Optical Single Crystal
Info: 9526 words (38 pages) Dissertation
Published: 2nd Feb 2022
Tagged: Chemistry
Growth, Spectral, nonlinear and DFT computations of a third order nonlinear optical single crystal: 2-amino 4-methyl pyridinium 4-methoxy benzoate (2A4MP4MB)
Abstract
In this work, single crystals of organic charge transfer complex, 2-amino-4-methyl pyridinium-4-methoxy benzoate (2A4MP4MB) was grown by controlled slow evaporation solution growth technique using ethanol as a solvent at room temperature. Single crystal XRD analysis confirmed the crystal system and lattice parameters of 2A4MP4MB. The crystalline nature, presence of various vibrational modes and other chemical bonds in the compound have been recognized and confirmed by powder X-ray diffraction, FT-IR and FT-Raman spectroscopic techniques respectively.
The presence of various proton and carbon positions in title compound was confirmed using 1H NMR and 13C NMR spectral studies. The wide optical operating window and cut-off wavelength were identified and bandgap value of the title compound was calculated using UV-Vis-NIR study. The specific heat capacity (cp) values of the title compound, 1.712 J g-1 K-1 at 300 K and 13.6 J g-1 K-1 at 433 K (melting point) were measured using Modulated Differential Scanning Calorimetric studies (MDSC).
From Z-scan study, non linear refractive index (n2), non linear absorption (β) and third order nonlinear susceptibility (χ (3)) values were determined. The Vickers micro hardness test was carried out at room temperature on crystallographic planes (100), (011) and (111) and obtained results were investigated using classical Meyer’s law. In addition, DFT calculations were carried out for the first time for this compound. These characterization studies performed on the title compound planned to probe the valuable and safe region of optical, thermal and mechanical properties to improve efficacy of 2A4MP4MB single crystals in optoelectronic device applications.
Keywords: Crystal growth, X-ray diffraction, Z-scan studies, Photoconductivity, Vickers micro hardness studies, DFT calculations.
1. Introduction
In recent past, many new organic crystals have been encounter based on the predictive molecular engineering approach and have been depicted to have potential applications in the fields of science and technology [1]. In recent years, research in the field of NLO single crystal design and development with advanced efficacy has been focused due to extensive range of applications like generation of higher harmonic frequencies, electro optic modulation, self focusing, frequency mixing, optical limiting, optical rectification, parametric oscillation, optical switching and terahertz wave generation etc [ ].
The development of highly efficient non linear optical materials for opto-electronic applications such as high speed information processing, optical communication, optical data storage have been the subject to intense research activity throughout the world. Organic nonlinear optical (NLO) single crystals acquire superior second and third order NLO properties compared to inorganic crystals, owing to advantages involve effortless tailoring of molecular arrangements, higher β values, low dielectric constant, multifunctional substitution, higher resistance to optical damage and maneuverability for certain device applications [2, 3, krish, thirupugal, brag].
Molecular flexibility of organic materials is an added advantage to enhance their nonlinear optical properties in a desired manner [4]. The various organic sub-networks induce noncentrosymmetry in the bulk and enhance the thermal and mechanical stabilities through hydrogen bonding interactions [5,6, brag]. An organic molecule with significant nonlinear optical activity generally consists of π electron conjugated moiety substituted by an electron donor group on one end of the conjugated structure and an electron acceptor group on the other end [10].
The conjugated π electron moiety provides a pathway for the entire length of conjugation under the perturbation of an external field. The donor and acceptor groups provide the ground state charge asymmetry of the molecule, which is required for second order and third order nonlinearity [11-12]. In supramolecular architecture, hydrogen bonding arrangements formed between pyridine and carboxylic acid derivatives has been confirmed as an important organizing force [sarkar, ballabh].
2-amino-4-methyl-pyridine (2A4MP) is a pyridine based complex possessed nitrogen (amino group), which readily accepts proton and donating electrons in the route of assembly of donor-acceptor (D-A) system, detected in general with different kinds of organic acids like carboxylic, benzoic and other acid derivatives.
In addition, the nitrogen atom attached with pyridine ring of 2A4MP favored reactions with various organicacids during formation of salts. Recently, organic compounds such as 2-amino-4-methyl pyridinium-4-Nitro phenolate 4-Nitrophenol [18], 2-amino-4-picolinium 4-aminobenzoate [20], 4-dimethyl amino–N-methyl-4-stilbazolium tosylate (DAST) [4,5], 4-dimethylaminopyridinium dihydrogen phosphate [14], 2-amino pyridinium trichloro acetate [13], Dimethyl amino pyridinium-4-Nitrophenolate 4-Nitrophenol (DMAPNP) [17] 2,6-Diaminopyridinium-4-Nitrophenolate 4-Nitrophenol (DAPNP) [16], 2-Aminopyridinium-4-Nitrophenolate Nitrophenol (2APNP) [15], 2-amino-5-chloro pyridinium-L-Tartrate [ ], 2-amino-5-chloro pyridinium-4-amino benzoate [ ] have been reported to show the evidence of significant NLO properties.
However, only a preferred group of compounds listed above acquired large molecular polarizabilities owing to their favorable delocalized electron (π –electrons) arrangements with improved secondand thirdorder optical nonlinearities, exhibited enhanced physicochemical properties.
In this direction, an organic nonlinear optical material 2-amino-4-methyl pyridinium-4-methoxy benzoate (2A4MP4MB) has been synthesized incorporating 2-amino-4-methyl pyridine (2A4MP) and 4-methoxy benzoic (4MB) acid.
A detailed literature survey showed there were no other studies available in this material. Based on these facts in the present work, 2-amino 4-methyl pyridinium 4-methoxy benzoate (2A4MP4MB) single crystals were grown by slow evaporation solution growth technique and the physiochemical properties such as single crystal XRD, powder XRD, FTIR, FT-Raman, NMR studies have been carried out.
Further optical (both linear and nonlinear) studies like UV-VIS-NIR, Photoluminescence (PL) spectroscopy, photoconductivity, Laser Damage Threshold (LDT) and Z-scan studies were reported for the first time. In addition, quantum chemical density functional calculations (DFT) including Natural bond orbital (NBO) analysis, Frontier Molecular Orbital (FMO) analysis, Mulliken atomic charge studies, Molecular electrostatic potential have been calculated for the 2A4MP4MB crystal.
2. Experimental Details
2.1 Synthesis, microanalysis and growth of 2A4MP4MB
Commercially available 2-amino 4-methyl pyridine (C6H8N2) (AR grade) and 4-Methoxy benzoic acid (C8H8O3)(AR grade) were purchased from Sigma Aldrich and used without further purification. The title compound salt was obtained by adding one mole of 2-amino 4-methyl pyridine and one mole of 4-methoxy benzoic acid in methanol solvent. The two solutions were mixed together and stirred well for about 2 hours to get homogeneous solution using mechanical stirrer and resulting solution was filtered through Whatmann 40 filter paper.
The reaction mechanism of 2A4MP4MB is shown in scheme 1. The Carbon-Hydrogen-Nitrogen (CHN) elemental composition percentage of 2A4MP4MB crystals were determined using Vario EL III Elemental analyzer (Germany) employing helium as a carrier gas. The analyzed results were given in Table 1. From the table, it could be observed that the experimentally obtained and theoretical results closely agreed with each other, also confirmed the presence of the compound.
The crystal growth vessel having filtrate was then covered using very thin polythene sheet to avoid fast evaporation. For controlled growth of 2A4MP4MB single crystal, the crystal growth vessel containing filtrate was placed in a constant temperature bath, whose temperature could be controlled by a Programmable temperature controller (Model: 3216; accuracy ± 0.01 °C). For the growth of single crystals of the title compound, slow evaporation solution growth method was employed and HPLC grade methanol was used as a solvent. The grown single crystals present inside vessel were taken out cautiously with the help of cleaned forceps.
The typical dimension of crystal was about 10 x 6x 4 mm3 collected, dried and then used in X-ray diffraction studies. The Theoretical BFDH morphology of the crystalline compound was resolved with the help of unit cell and positional coordinates. Fig. 1(a) and Fig. 1(b) showed as grown single crystal and theoretical BFDH morphology of the title compound.
2.2 Solubility studies
The variation in solubility of title compound as a function of temperature is shown in Fig. 2.The information on solubility and Metastable Zone width (MSZW) plot areusefultoascertainoptimal growth and perfection in crystallization process. The solubility of a material should be in a controlled level to enhance the growth of superior quality single crystals. Further, it is required that thesolute should remain in the solution till higher level of supersaturation has been attained in the solution to promote spontaneous nucleation.
The solubility data of 2A4MP4MB was measured using methanol solvent as a function of temperature (from 25 °C to 45 °C) in the interval of 5 °C. Initially, the saturated solution of the compound has been prepared in a well-controlled thermal environment. The solution with excess amount of solute in the beaker was allowed to agitate for nearly 5 hours before each sample was taken out. Solubility of the material was then measured gravimetrically.
To prepare homogeneous mixture, solution in the vessel was filtered and then it was preheated to 5 °C above its saturated temperature. Then the solution was maintained at same temperature for 2 hours before cooling process was initiated. The equilibrium-saturated solution in the growth vessel was then cooled from its overheated temperature (at a cooling rate of 2 °C/ h) until afirst visible crystal was observed. The saturated and nucleation temperature difference was taken as metastable zone width (MSZW) of the system [28]. The solubility of the material was found to rise almost linearly with the temperature.
2.3 Characterization details
The single crystal sample obtained from spontaneous nucleation process was subjected to single crystal X-Ray Diffraction method with the help of Bruker smart Apex CCD diffractometer with graphite monochromated MoKα radiation ( = 0.71073 Å) in the 2θ range 10–79.96o at a scan rate of 0.05o /sec, at ambient temperature using ω-scan method. The vibrational spectroscopic characterizations were recorded using Fourier Transform Infrared Spectroscopy (FT-IR) and FT-Raman spectra of the synthesized material in the wave number range 400–4000 cm-1. FT-IR spectrum of pelleted 2A4MP4MB was recorded using JASCO spectrometer (Model: 1410) in the range 4000-400 cm-1 with resolution 4 cm-1 and scanning speed of 2 mm/s. Whereas, FT-Raman spectrum was recorded using computer interfaced Bruker RFS 27 standalone FT-Raman spectrometer in the wave number range of 50-4000 cm-1.
The 400 MHz proton (1H) and carbon (13C) NMR spectral studies were carried out using Bruker NMR spectrometer by dissolving the powdered polycrystalline 2A4MP4MB in DMSO solvent at 293 K. The UV–Vis transmission spectrum of 2A4MP4MB crystal was recorded using the JASCO V-630 model spectrophotometer (wavelength accuracy ±0.2 nm). Whereas, Jobin Yvon Fluorometer FL3-11 was employed to record emission and absorption spectra of the compound at room temperature.
The refractive index measurement of title compound was carried out using Metricon model 2010/M at wavelength of 632 nm. The laser damage threshold study of title compound was carried out using Q-switched Nd:YAG laser with pulse width of 10 ns, frequency of 10 Hz and focal length of 40 cm. To carry out laser damage studies, freshly made flat (1 0 0) plane 2A4MP4MB single crystals with typical dimensions of 9-10× 7-8 × 4-5 mm3 were carefully polished using silk cloth containing alumina very finely dispersed in methanol solvent, was employed.
In this method, laser beam was focused either vertically or horizontally on the sample, at a distance of 300 µm between each focusing point. For every exposure, energy of the beam was increased by 50 mJ. When damage occurred, the beam energy and diameter of the spot were recorded.
Third order nonlinear optical properties like nonlinear refractive index and nonlinear absorption were examined using Z-scan technique. The Z-scan studies were performed using continuous He-Ne laser beam (λ= 632.8 nm) as an excitation source and it was focused using a convex lens with focal length of about 3.5 cm. In this study, the crystal sample was translated across the focal region (+z to –z) along the axial direction of a focused laser beam. In open aperture Z-scan, intensity dependent absorption was obtained without placing an aperture at the detector to get data about nonlinear absorption (β). In closed aperture method, transmitted energy through the sample is measured with the aperture placed at far field position for computing nonlinear refractive index (n2). A photo-detector through aperture collected focused laser beam transmitted through the sample and measured its intensity with the help of a digital power meter.
The photoconductivity behavior of 2A4MP4MB single crystal was examined using HHVc cryostat (model-CMT-25) with Keithley source meter (model-6487 Picometer).The photoconductivity study was carried out using well-polished (002) plane of suitably cut and polished 2A4MP4MB single crystal. A Halogen lamp employed with tungsten filament acts as a source for the photocurrent measurement via KEITHLEY 6487 Picoammeter. Dark current values were recorded for the sample with electric field values ranging from 0 to 60V. However, Photocurrent values were recorded for the sample, when it is exposed to the illumination of 50W halogen lamp used with iodine vapour.
Hardness studies for the grown crystals were carried out using LEITZ WETZLAR instrument attached with Vicker’s hardness pyramidal indenter to an incident light microscope. In the measurement of hardness of 2A4MP4MB, crack and inclusion free single crystals of typical size of 12×10×8 mm3 (l×b×h) were selected. The volume and surface area of the sample measured are 0.891 cm3 and 6.391 cm2 respectively. The grown crystal was properly mounted on the base of the microscope and the indentations were made on the surface by varying the applied load from 10g to 100g. Five indentations were made for each load and the average diagonal lengths of the indented impressions were measured using an optical microscope.
2.4 Computational details
Quantum chemical calculations of 2A4MP4MB were carried out using Gaussian03 software [22] program and also by the Gauss View [23] molecular visualization program. From B3LYP ( Becke’s three-parameter hybrid model employing Lee-Yang Parr correlation functional) method which consist of the Lee–Yang–Parr correlation functional with hybrid exchange functional was first proposed by Becke, Natural bonding orbital (NBO) analysis, Frontier molecular orbital (FMO) analysis, Molecular Electrostatic Potential and Mulliken atomic charge values have been calculated for the 2A4MP4MB crystal using DFT/B3LYP-6-311++G (d,p) level of theory [24-27].
The occupancy of donor and acceptor levels and the stabilization energy corresponding to different overlapping energy levels in title compound were investigated using B3LYP/6-31G basis set. Also, the shapes of HOMO and LUMO orbital and the Molecular Electrostatic potential in 3D (3-dimensional) of the title compound were plotted at B3LYP/6-31G level.
3. Results and Discussion
3.1 Elemental analysis
The elemental compositional percentage of 2A4MP4MB crystals was determined and results were given in Table 1. Experimental carbon, hydrogen and nitrogen percentages agreed with the theoretically obtained values, which confirmed the presence of compound.
3.2 Single crystal and Powder X-ray diffraction studies
The 2A4MP4MB single crystal was found to crystallize with centrosymmetric space group P21/c, which is an essential requirement for third harmonic generation. The cell parameters of 2A4MP4MB single crystal were measured to be a = 10.330(2) Å, b = 11.712(2) Å, c = 11.631(2) Å and α= 90°, β= 105.53(2)°, = 90° and the unit cell volume is equal to 1285.38 Å3 and the number of molecules present in unit cell (Z) was 4. The results obtained were spotted to agree well with those of the reported data [galle ramon]. The comparison of obtained single crystal X-ray diffraction (SXRD) values with thereported data values is shown in Table. 2. Moreover, the obtained crystal data established the event of protonation in the 2-amino-4-methyl pyridinium-4-methoxy benzoate molecule. Fig. 3 showed the molecular packing diagram of 2A4MP4MB.
Fig. 4(a) showed the sharp peak intensities of powder X-ray diffraction (PXRD) pattern for 2A4MP4MB and the PXRD obtained with the help of MERCURY software is shown in Fig. 4(b). The planes corresponding to different reflections in PXRD pattern were also indexed using the same software [35]. From experimental and simulated PXRD patterns, it could be observed that both were in close agreement with each other. These informations will be helpful for finding the crystal phase using simple PXRD pattern rather than repeating the SXRD characterization studies.
3.3 Optimized molecular geometry
Optimized single crystal molecular geometry computations were carried out for the title compound using Density functional theory (DFT) analysis at B3LYP level, with the help of split valence basis set 6-311++ (d, p) without incorporating any constraint to potential energy surface. The optimized molecular geometry along with the IUPAC numbering scheme of 2A4MP4MB molecule is shown in Fig. 5. The data of structural parameters obtained by ab initio method for 2A5CPLTA are reported and compared with single crystal XRD data values in Table. 3.
From the computed values, it was found that most of the optimized bond lengthsare slightly greater than the experimental values which could be attributed to the fact that the theoretical calculations were performed for isolated gaseous phase molecules whereas the experimental results were obtained for solid phase. It may be pointed out that the computed data values with the aid of B3LYP method correlate satisfactorily with the observed experimental data.
The carbon-carbon bonds in benzene ring were not of equal length due to the presence of nitrogen in the ring. However, the differences between the six C-C bond distances were small. The longest bond distance in C(1) – C(2) (optimized data) compared with other C-C bond distances could be due to the fusion of nitrogen atom at these carbons. Comparing the bond distances of the hetero aromatic ring, all differ significantly from each other owing to the dissimilarity in electro negativities of the linked atoms. The C(4)-Cl(7) (Optimized structure atoms) bond distance is the longest (1.7297 Å) while the C(2)-H(9) is the shortest (0.938 Å). The longest C(4)-Cl(7) distance attributes the pure single bond character. With electron donating substituents presence in benzene ring, the ring symmetry is distorted, thereby giving ring angles lesser than 120° at the point of substitution it was slightly larger than 120° at ortho and meta positions.
The variation in bond angle values relies on the electronegativity of central atom, presence of lone electron pair and conjugation of double bonds. The decrease in bond angle is observed due to the decrease in electronegativity values of the central atom. Consequently the bond angle C(5)-N(6)-C(1) is 123.73°. The 2A5CPLTA is a compound in which three different substituents [chlorine atom (-cl), amino group (-NH2) and Nitrogen atom] in the benzene ring and the fourth substituent (L-tartrate anion) which is in contact with 2-amino-5-chloro-pyridinium cation through hydrogen bond. The pyridinium ring appears distorted owing to these substituents and it is evident from the bond lengths C(1) – C(2) ( ~1.376 Å), C(1) – C(5) (~1.4094 Å), C(3) – C(4) (~1.4205 Å) when compared with the bond length C4-C5 (~1.400 Å). The hybridization owing to the substituent atoms attached with carbon atoms in ring resulted in the distortion in bond lengths.
From DFT calculations, it became apparent that the decreases in bond angle values by 8° were observed in C6-C7-O3 and C7-C8-O4 from 120°. In C8-C9-O5 the lowering of bond angle by 5.5° from 120° was also observed. The enhancement of bond angle was also observed in C8-C9-O6 by 2.7° from 120°. The enhancement of angles in tartrate anion was due to the attraction between lone electron of O22 and hydrogen in N6 of pyridinium ring. It was also observed that the optimized values were in good agreement with the experimental values.
The optimized and experimental results were observed to agree well for the torsional angles C2-C1-N8-H13, N6-C1-N8-H13, N6-C1-N8-H13, N6-C1-N8-H13, N6-C1-N8-H14. The differences in torsional angles between the experimental and optimized results might be due to the intermolecular bonding between 2-amino-5-chloro pyridinium cation and L-tartrate anion. Table. 4 showed Fractional atomic coordinates, equivalent isotropic displacement parameters (Å2) of 2A5CPLTA single crystal.
NMR spectral studies
1H NMR spectrum
Proton NMR spectroscopy, commonly accepted as Proton Magnetic Resonance (PMR), is an important component in the structural evidence of a synthesized compound and it shows relationship between various signals in spectrum and relationship between the various types of hydrogen atoms in the molecules. The proton and carbon NMR spectrum were carried out by dissolving powdered 2A4MP4MB crystals in DMSO solvent at temperature of about 293 K. It is an effective tool used to show the ratio of signals having same base areas caused due to the existence of number of hydrogen atoms.
PMR revealed that the dependence of splitting of a principal signal in to doublet or triplet or quartet signal on the presence of nearby unlike protons [15, 21, 22]. It is implicit from PMR that molecular structure determination is an easy process in simple compounds all the way through spin-spin splitting pattern using n+1 rule, here n is assigned as number of protons available in nearby carbon atom. It gives an idea about shift in the location of spectral signals corresponding to hydrogen atoms present in various chemical surroundings with that of several randomly selected standards. Furthermore, chemical shift takes place between atoms within the sequence of molecules relies on electron and molecular situation with in the nuclei.
The 1H NMR spectrum of the compound is shown in Fig. 4(a). The signal appeared at δ=2.16 ppm was assigned to a methyl proton in the 7’ position of 2-amino 4-methyl pyridinium cation of the title compound. The three protons connected in 7th position of 4-methoxy benzoate anion was attributed at δ=3.83 ppm. The broad signal appeared at δ=5.52 ppm was attributed to proton in the amine group (2’ position) of 2-amino 4-methyl pyridinium ring. The two signals emerged at δ= 7.01 and 7.03 were attributed to CH protons in 3 and 5 positions of 4-methoxy benzoate anion. The signals at δ=7.78 ppm and 7.801 ppm were attributed to CH protons in 2 and 6 positions of 4-methoxy benzoate ring. The peak appeared in the spectrum at δ=7.8ppm was attributed to proton joined with carbon atom in 3’ position of 2-amino 4-methyl pyridinium ring. The peak appeared at δ=8 ppm was attributed to the proton attached with carbon in 6’ position of 2-amino- 4-methyl pyridinium ring of the title compound.
Fig. 4(a). 1H NMR spectrum of 2A4MP4MBA
13C NMR spectrum
The 13C NMR spectrum of the compound is shown in Fig. 4(b). In 13C NMR of the title compound the signals owing to various carbon atoms were marked in the chemical structure. In spectrum signals appears at δ=20.9 and 56 ppm were attributed to carbon atoms in the methyl groups of 2-amino 4-methyl pyridinium ring and 4 methoxy benzoate respectively. The doublet appeared at δ=113.12 was assigned to carbon atoms in 3 and 5 positions of 4 methoxy benzoate ring. The 2- amino 4-methyl pyridinium ring carbon atoms in positions 3’, 5’ and 2’, 6’ yielded their signals at δ=129.89 and 144.98 ppm respectively shown in Fig. 4(b). Carbon atoms in 2 and 6 positions of methoxy benzoate yielded equivalent signals at δ=130.08 ppm. The methyl carbon atom in position 7’ of 2-amino 4-methyl pyridinium yielded single peak signal at δ=147.9 ppm. The signal appeared at δ=167.8 ppm was attributed to C=C between carbon atoms in 3 and 4 positions of 4 methoxy benzoate ring. The single peak signal appeared at δ=190 ppm was attributed to (C=O) carbon atom in 7th position of 4-methoxy benzoate ring. Thus the presence of peaks in the NMR spectra confirms the presence of expected compound and also reveals the proper completion of the reaction [23].
Fig. 4(b). 13C NMR spectrum of 2A4MP4MB
FTIR spectral studies
FTIR spectrum of 2A4MP4MB crystal was recorded using PerkinElmer spectrometer in the range of 4000-400cm-1 by employing KBr pellet method. The FTIR spectrum of the compound is shown in Fig. 5. The FTIR spectrum frequency assignments were carried out with the help of standard spectra of functional groups [24, 25].
The FTIR spectrum of 2A4MP4MB helps to validate the existence of functional groups in grown crystal. The peak signal detected at 3253 cm-1is assigned to the presence of OH group. C-H asymmetric and symmetric stretching vibrations of methyl groups in the compound give raise to peak signals at 2996 and 2837 cm-1 respectively. Two weak absorption peaks observed at
2757.7 cm-1and 2837 were attributed to O=C-H stretching vibrations in 4-methoxy benzoate ring. The peak signal in spectra emerged at 2555 is due to overtone or combination modes of COO– group. The signals at 2027 and 1948 cm-1 were attributed to combination and overtone of NH group. The COO– asymmetric stretching vibration is observed at 1676 cm-1. Aromatic rings due to their ring skeletal vibrations move up their peaks at 1600, 1502, 1455 cm-1.The absorption at 1502 cm-1is characteristics of N-C-N vibration. The OH bending vibration is found at 1385 cm-1. Peak signal at 1306 cm-1 corresponds to CH2 twisting mode of vibration. The C-O-H in-plane bending vibration is observed at 1247 cm-1 .
The peak at 1105 cm-1 is assigned to C-C-O stretching vibration. The C-H in-plane bending vibration is observed at 1031 cm-1. The absorption at 977 cm-1 is due to C-C stretching vibration. The C-O-H out of plane bending vibration is observed at 930 cm-1. The 1,4-disubstitute aromatic ring vibrations give rise to out plane C-H bending vibration at 846 cm-1. The peak at 787 cm-1 is characteristics of CH2 rocking vibration. C-H bending mode vibration results a peak at 748 cm-1. The O-C-O bending vibration is observed at 695 cm-1. The peak at 611 cm-1 is due to the OH bending vibration. The C-O bending vibration is observed at 507 cm-1. C-C-C-C in-plane bending vibration showed its peak at 427 cm-1 in the spectrum.
Fig. 5. FTIR spectrum of 2A4MP4MB
Raman spectral analysis
The Raman spectrum of the compound was recorded using Nicolet DXR Smart Raman spectrometer is shown in Fig. 6. Peak identification and assignments were carried out using standard spectral data is shown in Fig. 6 [26]. The peak at 3066 cm-1 is due to the OH asymmetric stretching vibration. The NH asymmetric stretching vibration is observed at 2999 cm-1. The peak at 2924 cm-1 is assigned to CH2 asymmetric stretching vibration. The CH asymmetric stretching vibration is found at 2836 cm-1. The peak at 1603cm-1 is characteristics of NH deformation mode.
The COO asymmetric stretching vibration is observed at 1575 cm-1. The peak at 1492 cm-1 is due to CN asymmetric stretching vibration. The CN symmetric stretching vibration is found at 1452 cm-1. The peak at 1310 cm-1 is assigned to CH2 scissoring mode of vibration. The CH2 wagging vibration is observed at 1249 cm-1. The peak at 1139 cm-1 is due to NH2 rocking vibration. The peak found at 1105 cm-1 is assigned to CH2 wagging vibration. The C-C stretching vibration is observed at 987 cm-1. The peak at 852 cm-1 is due to the CH2 rocking vibration is observed at 786 and 762 cm-1. The peak at 635 cm-1 is due to O-C=O stretching vibration. The COO- wagging vibration gives its peak at 570 and 520 cm-1. The peak at 459 cm-1 is assigned to C-N wagging vibration and C-C-C-C in-plane bending vibration observed at 431cm-1. The internal vibration of the molecule is observed at 363 and 308 cm-1.
Fig. 6. FT-Raman spectrum of 2A4MP4MB
UV-visible spectral analysis
In order to study linear optical characteristics, grown crystals of 2A4MP4MB with thickness about 2 mm were subjected to UV-Vis-NIR spectral analysis in the wavelength range from 200 to 1200 nm, using Varian Cary UV-Vis-NIR spectrophotometer is shown in the Fig 7. In NLO crystalline compounds, improved optical transmittance is one of the most important properties. Owing to occurrence of absorption peaks in between shorter to longer wavelength region in a material, conversion efficiency of wavelength get altered. Hence it is essential to monitor absorptions in the above said range.
From the spectrum (Fig. 7), it is clear that UV cut-off wavelength of grown 2A4MP4MB crystal was noted at 353 nm. The crystal is entirely transparent beyond cut-off wavelength up to 1200 nm. Due to the electronic transition between ‘non-bonding’ n orbital and anti-bonding π orbital represented as π*, strong absorption peak has taken place around 353 nm [27]. Moreover transparent character of crystal is suitable and prerequisite for second and third harmonic lights with wavelengths (λ = 532 nm) and (λ = 354.6 nm) respectively originated from Nd : YAG laser with laser light wavelength (λ = 1,064 nm) [28]. Quantum manipulations were employed to determine optical absorption coefficient for fundamental absorption in grown 2A4MP4MB crystal. Since quantum manipulations using quantum perturbation theory to treat the incident radiation as quantum perturbation and couples valence band energy levels with the energy levels in conduction band.
The optical absorption co-efficient (α) of grown 2A4MP4MB crystal was obtained from the following relation
α= 2.303log (1/ T)d
Where,
d is the thickness of the crystal and
T is the transmittance.
The absorption co-efficient (α) of 2A4MP4MB was determined using the following relation.
(αhν)= A (hν-Eg)n
Where A is constant. ‘h’ is Planck’s constant and
νis frequency of incident photons. The above equation is well suitable for allowable direct transition between the simple parabolic bands [29]. The optical energy gap (Eg) of 2A4MP4MB is calculated using Tauc’s plot of
hνversus (αhν) 1/n, by extrapolating the linear portion of the curve near start of absorption edge towards the energy axis is shown in Fig. 8. The band gap value of 2A4MP4MB crystal was estimated at 3.61 eV. The wide band gap of this crystal shows large transmittance of visible region.
Fig. 7. Opticaltransmission spectrum of 2A4MP4MB
Fig. 8. Tauc’s Plot of 2A4MP4MB
Photoluminescence studies
Photoluminescence (PL) spectroscopy is a significant method to evaluate the defects and optical properties of the grown crystal as a photonic material. The existence of different energy states between valance band and conduction band are responsible for radioactive recombination, which can be obtained from photoluminescence (PL) spectrum. The luminescence of organic compounds essentially based on localized -electron systems within individual organic molecules. In general, PL signal depends on the density of photo excited electrons, intensity of the incident beam and also change with excitation position and wavelength. The emission spectrum of as grown 2A4MP4MB was carried out at room temperature using Varian Cary Eclipse Fluorescence Spectrophotometer.
By exciting the sample using Xenon lamp at 280 nm, the emission output was given to a monochromator. Emission intensity was recorded as a function of wavelength. The excitation spectrum is recorded in the range of 240-280 nm as shown in Fig. 9 reveals that the sample has been excited at the wavelength of 280 nm. (Fig. 10) shows the PL emission spectrum of 2A4MP4MB crystal. From the (Fig. 10.) it is obvious that a strong emission peak was observed at 330 nm, which could be attributed to intramolecular charge transfer between oxygen atom of carboxylate group and hydrogen atom of NH2 attached with 2-amino-4-methyl pyridinium ring [Thirumurugan]. The obtained result showed the title compound is an improved ultraviolet wavelength emitting material and also potential candidate for nonlinear optical applications. Therefore, the title compound crystals could be useful as a potential candidate in UV light emitting diode applications [30].
Fig. 9. Excitation curve of 2A4MP4MB
Fig. 10. Emission spectrum of 2A4MP4MB
Natural bond orbital analysis (NBO)
Natural bond orbital analysis gives the most accurate possible natural Lewis structure of molecule because all the orbital are mathematically chosen to include the highest possible percentage of the electron density. The NBO analysis will explain the interaction between both the filled and virtual orbital space information. it is also helpful in the investigation of intra and inter molecular interaction. The second order Fock matrix was carried out to evaluate the donors (i), acceptors (j) and their levels. The result of interaction is a loss of occupancy from the concentration of electron NBO of the idealized Lewis structure into an empty non-Lewis orbital. For each donor (i) and acceptor (j), the stabilization energy E(2) associates with the delocalization i → j is estimated as
E(2)= -qi(Fij)2j-i
where qi is the donor orbital occupancy, εi and εj are diagonal elements and F(i,j) is the off diagonal NBO Fock matrix element. NBO analysis gives an efficient scheme for interaction among bonds, and it also delivers a convenient basis for investigating charge transfer or conjugative interaction in molecular systems. The second order micro-disturbance theory were reported already for studying donor orbital, acceptor orbital and the interaction stabilization energy [31]. The more intensive interaction between electron donors and acceptors, i.e., the higher donation tendency from electron donors to electron acceptors leading to the larger stabilization energy.
Delocalization of electron density between occupied Lewis-type (bond or lone pair) NBO orbital and formally unoccupied (anti bond or Rydberg) non-Lewis NBO orbital correspond to a stabilizing donor–acceptor interaction. Among those interactions, It is pointed out that the stabilization energy corresponding to the overlap between LP(3) of O2 with anti-bonding LP(1) of C11 is highest than any other corresponding overlap of LP(3) of O3 with anti-bonding LP(1) of C14. The stabilization energy of the former overlap is 198.24kcal/mol. The transitions with larger delocalization energies are listed in Table 2.
| Donor(i) | Type | Occupancy | Acceptor(j) | Type | Occupancy | E2 (kcal/mol) | Ej –Ei (a.u.) | F(ij)(a.u.) |
| C3 – C4 | | 1.64994 | C11 | LP*(1) | 0.80032 | 39.58 | 0.14 | 0.075 |
| C3 – C4 | | C7 – C13 | * | 0.29915 | 22.62 | 0.28 | 0.072 | |
| C3 – C4 | | C9 – C12 | * | 0.40777 | 18.33 | 0.26 | 0.062 | |
| C4 – C9 | | 1.97105 | O6 – C12 | * | 0.03065 | 5.59 | 1.01 | 0.067 |
| C7 – C13 | | 1.69815 | C3 – C4 | * | 0.34401 | 16.83 | 0.29 | 0.063 |
| C7 – C13 | | C9 – C12 | * | 0.40777 | 24.55 | 0.27 | 0.074 | |
| C9 – C12 | | 1.67328 | C3 – C4 | * | 0.34401 | 22.07 | 0.31 | 0.074 |
| C9 – C12 | | C7 – C13 | * | 0.29915 | 15.71 | 0.3 | 0.062 | |
| O1 | LP (1) | 1.96331 | C11 | RY*(1) | 0.02025 | 7.34 | 1.47 | 0.093 |
| O1 | LP (1) | C11 | RY*(2) | 0.01044 | 3.52 | 1.39 | 0.063 | |
| O1 | LP (1) | O2 – C11 | * | 0.04717 | 6.7 | 1.18 | 0.08 | |
| O1 | LP (2) | 1.87603 | O2 – C11 | * | 0.04717 | 12.18 | 0.82 | 0.091 |
| O1 | LP (2) | C3 – C11 | * | 0.08069 | 16.03 | 0.72 | 0.097 | |
| O1 | LP (3) | 1.62913 | C11 | LP*(1) | 0.80032 | 183.58 | 0.13 | 0.154 |
| O2 | LP (1) | 1.95876 | C11 | RY*(1) | 0.02025 | 8.93 | 1.46 | 0.103 |
| O2 | LP (1) | O1 – C11 | * | 0.05159 | 6.66 | 1.14 | 0.078 | |
| O2 | LP (2) | 1.87472 | O1 – C11 | * | 0.05159 | 13.83 | 0.78 | 0.095 |
| O2 | LP (2) | C3 – C11 | * | 0.08069 | 17.57 | 0.72 | 0.101 | |
| O2 | LP (3) | 1.61111 | C11 | LP*(1) | 0.80032 | 198.24 | 0.13 | 0.16 |
| O6 | LP (2) | 1.85450 | C9 – C12 | * | 0.40777 | 29.87 | 0.33 | 0.095 |
| C11 | LP*(1) | 0.80032 | C3 – C 4 | * | 0.34401 | 31.91 | 0.15 | 0.081 |
| C9 – C12 | * | 0.40777 | C3 – C4 | * | 0.34401 | 113.39 | 0.03 | 0.081 |
| C9 – C12 | * | C7 – C13 | * | 0.29915 | 143.39 | 0.02 | 0.083 |
Table 2: Second order perturbation theory analysis of Fock matrix in NBO basis for 2A4MP4MB
Analysis of Frontier Molecular Orbitals (FMOs)
The highest occupied molecular orbitals (HOMOs) and the lowest-lying unoccupied molecular orbitals (LUMOs) are named as Frontier Molecular Orbitals (FMOs). The FMOs play a significant role in the optical and electric properties, quantum chemistry and UV–vis spectra. HOMO and LUMO energy characterizes the ability of electron accepting. While we are dealing with molecular orbitals interaction, the two that act together are commonly the highest energy occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the compound. These orbitals are a pair of orbitals in the compound, which allows them to interact more strongly. These orbitals are sometimes called the frontier orbitals, because they lie at the outermost boundaries of the electrons of compound. The frontier orbital gap supports to characterize the chemical reactivity and the kinetic stability of the molecule. A molecule with a small frontier orbital gap is generally associated with high chemical reactivity and low kinetic stability [32].
In addition, the FMOs are important in determining the ability of a molecule to absorb light. The calculated HOMO and LUMO energy values in gas phase are -0.28324 eV and 0.06941 eV respectively, and the frontier orbital energy gap value is 0.35265 eV. HOMO and LUMO energies and surfaces can be seen in Fig. 11.The narrow energy gap between HOMO and LUMO facilitates intramolecular charge transfer which makes the material to be NLO active.
Using HOMO and LUMO orbital energies, the ionization energy (I) and electron affinity (A) can be expressed as: I= -EHOMO and A= -ELUMO. From the value of ionization energy and electron affinity, the electronegativity ( = (I+A)/2), chemical hardness ( = (I-A)/2), Softness (S= 1/2) have also been calculated [33]. The electrophilicity index is also calculated by the expression = (-2/2) [34]. The calculated values of electronegativity, chemical potential, chemical hardness, softness, and electrophilicity index for the title molecule are 0.106915 eV, -0.14182 eV, 0.176325 eV, 2.835673 eV and 0.032414 eV. The hardness indicates the resistance to the deformation of electron cloud under small perturbation encountered through the chemical process. Soft systems are large and highly polarizable, while hard systems are relatively small and much less polarizable.
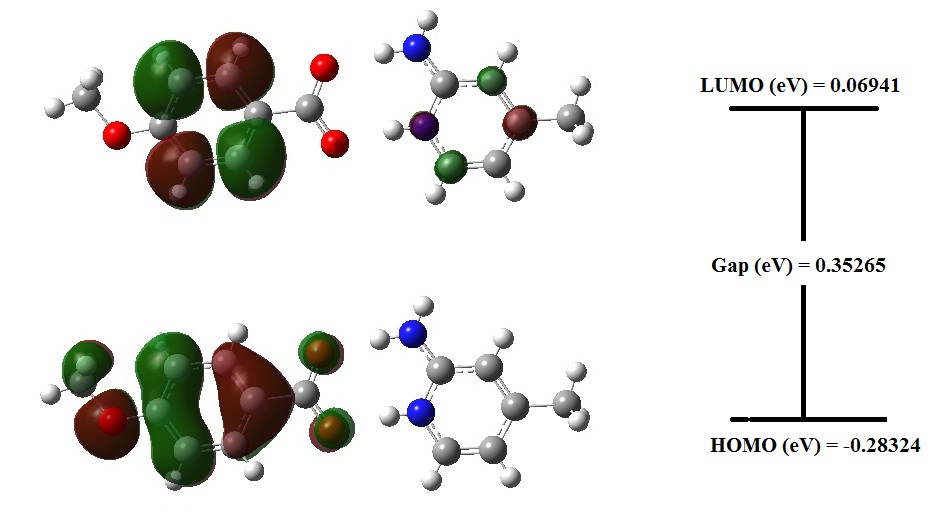
Fig. 11. Homo and Lumo plot of 2A4MP4MB at B3LYP/6-31+G.
Mulliken Atomic analysis
The calculation of atomic charges has a vital role in the application of quantum chemical calculation to molecular system, because the atomic charges affect many properties such as dipole moment, electronic structure, polarizability. The atomic charges of individual atoms and its total value of 2A4MP4MB molecule have been obtained by Mulliken population analysis and are depicted in Fig 12. From the figure, it could be observed that the atomic charges of 2A4MP4MB are ranging from -0.69422 and +0.58412. It could also be observed that all the oxygen atoms possess negative values whereas all the hydrogen atoms possess positive values. The variations in atomic charges of hydrogen atoms may be due to the formation of hydrogen bonding. However, the sum of Mulliken atomic charges of all atoms of 2A4MP4MB molecule becomes zero and maintains the charge neutrality. Mulliken population analysis is a good technique to explain differences in electro-negativities of atoms within the molecule with the support of MEP contour map. MEP and Mulliken population method can be used for calculating the reactive behaviour of chemical systems in both electrophilic and nucleophilic reactions.
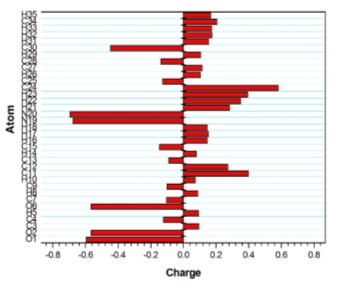
Fig. 12. Molecular Electrostatic Potential study
The MEP surface provides facts about the chemical reactivity of a molecule. The electrostatic potential generated in space around a molecule by the charge distribution is supportive to realize about the electrophilic or nucleophilic properties of molecule [35, 36]. In order to predict the reactive sites for electrophilic and nucleophilic attack, the molecular electrostatic potential in mesh form has been plotted for 2A4MP4MB and is shown in Fig. 13. In MEP map, different values of the electrostatic potential are represented by different colours: red and blue represents the regions of the most negative and positive electrostatic potential whereas green represents the region of zero potential. Intermediary colours represent intermediary electrostatic potentials. Potential increases in the order of: red
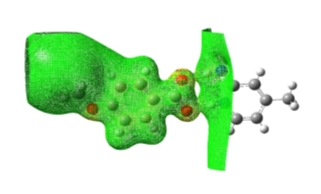
Fig. 13. Molecular electrostatic potential in mesh form.
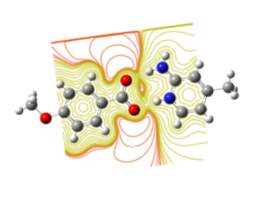
Fig. 14. Contour plot of energy surfaces of the title compound.
NLO effects
The first order hyperpolarizability and related properties (dipole moment, mean polarizability and anisotropy of polarizability) have been calculated by finite field approach, which is currently one of the procedures utilized for finding numerical NLO responses [37]. The components of βtot are defined as the coefficients in the Taylor series expansion of the energy in the external electric field. The Taylor series expansion is
E=E0-iFi- 12ijFiFj-16βijkFiFjFk-16γijklFiFjFkFl+…
Where Eo is the energy of the unperturbed molecule, Fi is the field at the origin, i, ij, βijk, γijkl are the components of dipole moment, polarizability, the first hyperpolarizabilities and the second hyperpolarizabilities, respectively. The total static dipole moment, mean polarizability, anisotropy of polarizability and first order hyperpolarizability can be calculated as:
= x2+x2+x2
= xx+yy+zz3
=2xx-yy2+yy- zz2+zz-xx2+6xx21/2
β=βxxx+βxyy+βxzz2+βyyy+βyzz+βyxx2+βzzz+βzxx+βzyy212
Since the values of polarizabilities and hyperpolarizabilities of Gaussian output are reported in atomic units (a.u.), the calculated values have been converted into electrostatic units (esu) for hyperpolarizability 1 a.u. = 8.6393 x 10-33 esu and polarizability 1 a.u. = 0.1482 x 10-24 esu. The calculated total static dipole moment, mean polarizability, anisotropy of polarizability and first order hyperpolarizability are 7.710516 Debye, 0.506Å esu, 0.864Å esu and 8.36954 x 10-32cm5 esu-1respectively. The first order hyperpolarizability of title compound is higher than that of urea (5.63 x 10-32cm5 esu-1). The compared values are listed in Table 3.
| 2A4MP4MB | Urea | |
| Total static dipole moment | 7.710516 | 4.302935 |
| Mean polarizability | 0.506 | 0.139 |
| Anisotropy of polarizability | 0.864 | 0.934 |
| First order hyperpolarizability | 8.36954 | 5.63 |
Table 3. Comparison of 2A4MP4MB values with urea
Conclusion
A single crystal of new nonlinear optical material 2-amino-4-methyl pyridinium-4-methoxy benzoate was synthesized slow evaporation solution growth method at ambient temperature. The grown crystal was bright, transparent and colorless crystal was obtained within 15-20 days with average dimension of 0.5 x 0.15x 0.2 cm3. The single crystal X-ray diffraction method was studied to determine the structure of the compound and shows that the compound crystallizes in monoclinic system. The powder X-ray diffraction method was studied to confirm the crystalline nature of the compound. The FTIR and Raman spectroscopic techniques were carried out to predict the various functional groups and other chemical bonds in the compound. The various kinds of protons and carbons in the compound were assigned through NMR spectral technique. UV-Vis absorption studies indicating the absence of absorption in the visible region, hence the crystal is expected to be transmittance in this region.
Photoluminescence study results revealed that the title crystal could be used as a potential candidate in NLO devices. Furthermore, Quantum chemical calculations showed different orbital shapes, 3D-Molecular electro static potential, Electric dipole moment, polarizability, first and second order hyperpolarizabilities of 2A4MP4MB crystal. Hence, all the above studies clearly indicate that the title compound could be used a better material in NLO devices.
References
[1] T. Malik, T. Kar, Mater. Lett. 61 (2007) 3826.
[2] Z.H. Sun, G.H. Zhang, X.Q. Wang, X.F. Chang, X.Y. Liu, Y. Fun, G. Yu, D. Xu, J. Cryst. Growth 310 (2008)2842.
[3] G. Shanmugam, K. Ravi Kumar, B. Sridhar, S. Brahadeeswaran, Mater. Res. Bull. 47 (2012) 2315.
[4] A. Datta, S.K. Pati, J.Chem. Phys, 118, 8420 (2003).
[5] A. Criado, M.J. Dianez, S.P.C. Garrido, I.M. Fernandes, M.E. Belsley, M. De Gomes 56 (2000) 888.
[6] J. Zaccaro, J.P. Sulvestrini, A.Ibanez, P.Ney, M.D. Fontana, J. Opt. Soc. 17 (2007)427.
[7] C.K. Lakshmana Perumal, A. Arulchakkaravarthi, P. Santhanaraghavan, P. Ramasamy, J.Cryst. Growth 241 (2002) 200.`
[8] G. Shanmugam, S. Brahadeeswaran, Spectrochimica Acta Part A 95 (2012) 177–183.
[9] G. Shanmugam, K. Thirupugalmani, V. Kannan, S. Brahadeeswaran, Spectrochimica Acta Part A 106 (2013) 175.
[10] L. Jothi, K. Ramamurthi, Indian J. Sci. Technol. 4 (2011) 6.
[11] T. Pal, T. Kar, Mat. Chem. Phys 91(2005) 343.
[12] P.V. Dhanaraj, N.P. Rajesh, G. Vinitha, G. Bhagavannarayana, Mater. Res. Bull. 46 (2011) 726.
[13] Gaëlle Ramon, Kate Davies and Luigi R. Nassimbeni, CrystEngComm, 16 (2014) 5802.
[14] J.I. Wu, R. Gopalakrishnan, C.ID. Tai, C.W. Law, Jpn. J. Appl. Phys. 43 (2004) 1507-1513.
[15] G. Shanmugam, S. Brahadeeswaran, Spectrochim. Acta Part A 95 (2012) 177-183.
[16] S. J. Kwon, M. Jazbinsek, O. P. Kwon, P. Gunter, Cryst. Growth Des. 10 (2010) 1552-1558.
[17] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,J.A. Montgomery Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar,J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A.Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa,M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox,H.P. Hratchian, J.B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann,O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K.Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S.Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K.Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J.Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L.Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M.Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, J.A.Pople, Gaussian 03, Revision D.01, Gaussian Inc., Wallingford, CT, 2004.
[18] L.J. Farrugia, J. Appl. Cryst. 30 (1997) 565–566.
[19] A. Frisch, A.B. Nielson, A.J. Holder, GAUSSVIEW User Manual, Gaussian Inc., Wallingford, CT, 2003.
[20] A.D. Becke, J. Chem. Phys. 98 (1993) 5648–5652.
[21] T.N. Mitchell, B. Costiella, NMR- From spectra to structures – An experimental approach, second ed., Springer, New York, 2007.
[21] Y. R. Sharma, Elementary Organic Spectroscopy Principles and Chemical applications, S.Chand & Co, New Delhi, 2004.
[22] P. Pandi et al. Spectrochim. Acta Part A 98 (2012) 7-13.
[22] R. M. Silverstein, G. C. Bassler, T.C. Morrill, Spectrometric Identification of Organic Compounds, Wiley, New York, 1981.
[23] G. Socrates, Infrared Characteristic Group Frequencies, Wiley- Interscience, Chichester, 1980.
[24] Norman B. Colthup, Lawrence H. Daly, Stephen E. Wiberley, Introduction to infrared and Raman spectroscopy, Academic Press, New York, 1964, pp 278.
[27] Sharma YR. Elementary organic spectroscopy. 4th ed. New Delhi: S. Chand; 2007
[28] Shanmugam G, Brahadeeswaran S. Spectroscopic, thermal and mechanical studies on 4-methylanilinium p-toluenesulfonate – anew organic NLO single crystal. Spectrochim Acta A, 95, 2012, 177–83.
[29] Tauc JC. Optical properties of solids. Amsterdam: North-Holland;1972. p. 372–5.
[30] M. Pope, C.E. Swenberg, Electronic Processes in Organic Crystals and Polymers,second ed., Oxford University Press, New York, 1999.
[31] L. Jun-na, C. Zhi-rang, Y. Shen-fang, Study on the prediction of visible absorption maxima ofazobenzene compounds, J. Zhejiag Univ. Sci. 6B (2005) 584.
[32] I. Fleming, Frontier Orbitals and Organic Chemical Reactions, Wiley, London,1976
[33] H. Chermette, J. Comput. Chem. 20 (1999) 129–154.
[34] R.G. Parr, W. Yang, J. Am. Chem. Soc. 106 (1984) 4049–4050.
[35] E. Scrocco, J. Tomasi, Adv. Quantum Chem. 11 (1978) 115–193.
[36] F.J. Luque, J.M. Lopez, M. Orozco, Theoret. Chem. Acc. 103 (2001) 343–345
[37] H.A. Kurtz, J.J.P. Stewart, K.M. Dieter, J. Comput. Chem. 11 (1990) 82–87.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Chemistry"
Chemistry is a science involving the study of the elements and matter at the atomic and molecular level including their composition, structure, properties, behaviour, and how they react or combine.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: