RIPK3 Blockade Attenuates Tubulointerstitial Fibrosis in a Mouse Model of Diabetic Nephropathy
Info: 6573 words (26 pages) Dissertation
Published: 25th Feb 2022
Tagged: Biomedical Science
Abstract
Receptor-interacting protein kinase-3 (RIPK3) is a multifunctional regulator of cell death and inflammation. RIPK3 controls cellular signalling through the toll-like receptor (TLR) pathway and the formation of the domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome, which have been well recognised to mediate renal fibrogenesis. The role of RIPK3 in diabetic kidney disease (DKD) induced renal fibrosis has not been previously determined. To define the action of RIPK3 in the development of DKD, wild-type (WT), RIPK3 -/-, endothelium-derived nitric oxide synthase (eNOS)-/- mice were induced to develop diabetes mellitus using multiple low doses of streptozotocin and maintained for 24 weeks. RIPK3 expression was upregulated and fibrotic responses were increased in the kidney cortex of mice with established diabetic nephropathy compared to control mice. Consistently, mRNA expression of inflammasome components, TLR4 and interleukin (IL)-1β as well as TGF-β1 and α smooth muscle actin (α-SMA) were increased in diabetic kidneys of WT mice compared to control mice. However, these markers were normalised or significantly reversed in kidneys of diabetic RIPK3 -/- mice. Renoprotection was also observed using the RIPK3 inhibitor dabrafenib in eNOS-/- diabetic mice as demonstrated by reduced collagen and fibronectin deposition and myofibroblast activation. These results suggest that RIPK3 is associated with the development of renal fibrosis in DKD due to downstream activation of the NLRP3 inflammasome and TLR4 and TGF1 signalling. Inhibition of RIPK3 results in renoprotection. Thus, RIPK3 may be a potential target for therapeutic intervention in patients with diabetic kidney disease.
Introduction
End-stage kidney disease (ESKD) is a major cause of morbidity and mortality in patients with diabetes mellitus. Renal fibrosis indicates progressive renal disease will ensue in most forms of chronic kidney disease (CKD). In recent years, the increasing prevalence of diabetes mellitus accounts for the majority of chronic kidney disease worldwide [1]. The mainstay of therapy for diabetic kidney disease (DKD) is currently limited to controlling blood glucose and blood pressure, generally with an agent that blocks the renin-angiotensin system [2]. To date, no specific therapy for preventing diabetic kidney disease is available. A successful continuum between innovative discovery science and rigorous translation of research findings is required to improve the outcomes of patients with diabetic kidney disease.
The receptor-interacting protein kinase (RIPK)3, a crucial kinase mediating necroptosis, has been implicated as a potential regulator of multiple inflammatory pathways, including toll-like receptor (TLR)4 [3] and domain-like receptor family pyrin domain-containing (NLRP) 3 inflammasome [4], which have been well established to promote tubular epithelial cell injury and interstitial fibrosis in various models of kidney disease, including DKD [5-10]. These studies suggest that RIPK3 may be a potential target to ameliorate DKD. Limiting the activation of the NLRP3 inflammasome and TLR4 by inhibiting RIPK3 has theoretical benefits in limiting renal fibrosis, including downstream effects on TGF1 expression and signalling. However, the function of RIPK3 in the development of DKD has not been elucidated.
Dabrafenib, known as a B-Raf inhibitor, suppresses the downstream Ras/Raf/extracellular-signal-regulated kinase (ERK)/ mitogen-activated protein kinase (MAPK) pathway [11] and has been approved by the U.S. Food and Drug Administration for the treatment of non-small cell lung cancer and melanoma expressing BRAFV600E mutations [12]. It is well documented that Ras/Raf/ERK/MAPK pathways are involved in renal fibrosis [13], suggesting dabrafenib may have renoprotective effects. Dabrafenib has also been reported to inhibit RIPK3 in mouse models of ischemic brain injury [14] and liver injury in human hepatocytes [15]. Compared to the other widely-used RIPK3 inhibitor GSK’872 [16, 17], dabrafenib shows more therapeutic potential for renal fibrosis because it has broader kinase inhibition. Given it is already in use in humans, repurposing for use in DKD is likely to have a facilitated route to market.
In this study, we examined the role of RIPK3 in DKD induced renal fibrosis using a streptozotocin (STZ)-induced diabetic mouse model. We found that RIPK3 deficiency attenuated diabetes-induced renal fibrosis, associated with reduced activation of the NLRP3 inflammasome, TLR4 and TGF1 signalling. Dabrafenib treatment also attenuated diabetes-induced renal interstitial extracellular matrix (ECM) deposition and myofibroblast activation. Our data support the tenet that RIPK3 may mediate diabetes-induced fibrosis through the NLRP3 inflammasome and TLR4 pathway. Hence strategies that include inhibition of RIPK3 may limit the development of diabetic kidney disease.
Methods
Human kidney biopsies
Human kidney biopsy specimens from patients with established diabetic nephropathy were kindly provided by the Department of Anatomical Pathology, Royal North Shore Hospital. Kidneys removed from patients, generally due to peripheral tumour but without known kidney disease, served as controls. This study was approved by the Human Research Ethics Committee of the Royal North Shore Hospital (HREC reference LNR/17/HAWKE/497).
Animal studies
6-8-week-old male wild-type (WT), RIPK3-/-, endothelium-derived nitric oxide synthase (eNOS)-/- mice (all C57BL/6 background) weighing 20-25g were used for this study. Mice were assigned to receive either 55 mg/kg STZ (Sigma-Aldrich) diluted in 0.1 M citrate buffer, pH 4.5, or citrate buffer alone by intraperitoneal injection for five consecutive days as described previously [18]. STZ-treated animals with blood glucose >16 mmol/L were considered as diabetic. eNOS-/- mice were used to study the effect of dabrafenib on the development of diabetic nephropathy. Alzet model 2006 osmotic pumps (0.15µl/hr of dabrafenib dissolved in vehicle dimethyl sulfoxide (DMSO) at a concentration of 16.7 mg/ml) were implanted in the subcutaneous tissues in the interscapular region of eNOS-/- diabetic mice to deliver dabrafenib or DMSO. Mice were weighed, and the blood glucose was monitored weekly with an ACCU-CHEK glucometer (Roche Diagnostics). Insulin (Lantus 2U/ day) was given subcutaneously if the blood glucose of mice exceeded 28 mmol or mice had significant weight loss (10-15% body weight loss in mouse with blood glucose >16 mmol/L). Mice were sacrificed 24 weeks after the induction of diabetes (as shown in Fig 5.1). All animals were housed in the Kearns Animal Facility of the Kolling Institute of Medical Research, with a stable environment maintained at 22 ± 1°C with a 2/12h light-dark cycle. Experiments were done in accordance with the National Health and Medical Research Council of Australia’s Code for the Care and Use of Animals for Scientific Purposes and were approved by the Animal Research Ethics Committee of the Royal North Shore Hospital (NSLHD reference RESP/15/206).
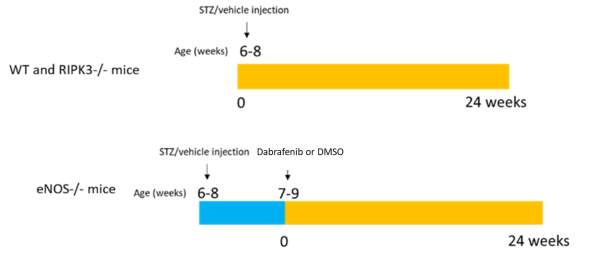
Figure 5.1 Timeline of DM model and dual strategies to assess the effect of limitation of RIPK3 on the development of diabetic kidney disease. Male mice at 6-8 weeks of age were induced to develop diabetes using a regimen of 5 doses of STZ as described above. After a week, eNOS-/- diabetic mice underwent surgery to implant the pump to deliver dabrafenib or vehicle. Mice were sacrificed 24 weeks after inducing diabetes.
RNA isolation and RT-PCR analysis
Total RNA was extracted from mouse kidney cortex using the RNeasy plus mini kit (Qiagen) by QIAcube (Qiagen). cDNA was synthesized with a cDNA synthesis kit (Roche). Predesigned probes (PrimeTime® Assay Std Probe 5′ 6-FAM™/ZEN™/3′ IB®FQ, Integrated DNA Technologies), including RIPK3 (Assay ID: Mm.PT.58.12712973.gs), Collagen IV ((Assay ID: Mm.PT.58.31838522), Fibronectin (Assay ID: Mm.PT.58.8135568), TGF-β1 (Assay ID: Mm.PT.58.11254750), -SMA (Assay ID: Mm.PT.58.16320644), MCP-1 (Assay ID: Mm.PT.58.42151692), F4/80 (Assay ID: Mm.PT.58.11087779), TLR4 (Assay ID: Mm.PT.58.41643680), NLRP3 (Assay ID: Mm.PT.58.13974318), ASC (Assay ID: Mm.PT.56a.42872867), IL-1 β (Assay ID: Mm.PT.58.41616450), Tubulin (Assay ID: Mm.PT.58.13069923) were measured. Tubulin was used as endogenous reference gene.
Histology and Immunohistochemistry
Kidney tissues were fixed in 4% paraformaldehyde and embedded with paraffin. 4 μm kidney sections were cut and stained as follows: After deparaffinisation with xylene, slides were immersed in decreasing concentration of ethanol (100%, 100%, 95%, 70%) and rinsed in a container of running water. Citrate buffer at pH 6 was heated to 99 ℃ for epitope retrieval. Slides were placed in hot retrieval buffer for 20 minutes incubation followed by 20 minutes cooling to room temperature. Slides were blocked for 15 minutes (protein block serum-free ready to use x0909, Dako). Immunohistochemical staining was processed in Thermo Scientific Sequenza system. Slide were incubated overnight at 4℃ with primary antibodies as follows: RIPK3 (Abcam: ab56164), type I collagen (Abcam, ab34710, 1:750), type III collagen (Abcam, ab7778, 1:500), type IV collagen (Abcam, ab6586, 1:1000), Fibronectin (Abcam, ab 45688, 1:750), a-SMA (Sigma-Aldrich, A2547, 1:10000), p-Smad2/3 (Santa Cruz Biotechnology, sc-11769R, 1:150). Slides were then washed with TBS-T (50mM Tris, 150mM NaCl, 0.05% Tween 20, pH 7.6), and incubated with associated secondary antibodies (EnVision+system-HRP labelled polymer anti-rabbit k4003, Dako) for 30 minutes at room temperature. After washing with TBS-T, kidney sections were covered with DAB (liquid DAB+substrate chromogen system k3468, Dako) for 10 minutes. All experiments included controls without primary antibody. Positive signals in the renal cortex were quantified using Image J software as previously described [19].
Statistical analysis
The results are presented as mean ± S.E.M. The differences between two groups were analysed by two-tailed t-tests, and comparison of multiple groups was analysed by one-way analysis of variance ANOVA followed by Tukey’s multiple comparisons test. The survival curves were analysed by Log-rank (Mantel-Cox) test. A two-sided p-value
Results
Upregulation of RIPK3 expression in the diabetic kidney
To determine whether RIPK3 is upregulated in diabetic kidney, RIPK3 protein expression was measured in kidney biopsies collected from normal nephrectomy specimens (Fig. 5.2a: Ctrl) and patients with established diabetic nephropathy (Fig. 5.2a: DN) using immunohistochemistry. RIPK3 protein was clearly upregulated in proximal tubules (black arrows) of diabetic kidneys compared to normal kidneys.
RIPK3 protein expression was examined using immunohistochemistry in kidneys of mice with or without diabetic nephropathy. Consistent upregulation of RIPK3 protein (black arrows) was observed in the kidneys of diabetic mice compared to control kidneys (Fig. 5.2b). mRNA expression of RIPK3 was assessed using RT-PCR. As shown in Fig. 5.2c, RIPK3 mRNA expression was significantly increased in diabetic mice compared to non-diabetic controls (P
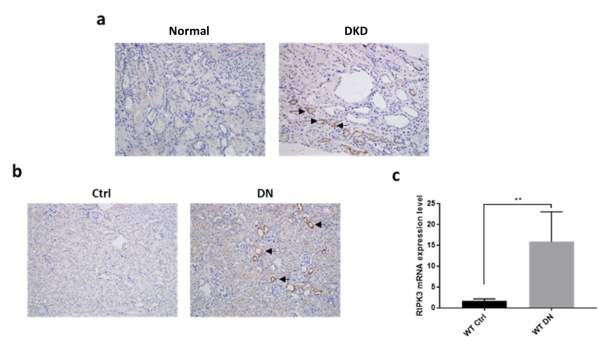
Figure 5.2 Increased receptor-interacting protein kinase (RIPK) 3 in diabetic kidney disease.
(a) Representative images of RIPK3 stained kidney biopsies collected from normal nephrectomy specimens and patients with established DKD. Magnification: 20×. (b) Representative images of RIPK3 stained kidney sections in mice (wild-type WT or RIPK3-/-) +/- diabetes mellitus. Magnification: 20×. (c) RIPK3 mRNA expression fold change was measured by RT-PCR in the same groups as in (b). Tubulin was used as reference gene. Statistical analysis was performed using two-tailed t-tests. Results are presented as mean ± S.E.M. **P
RIPK3 gene knockout reduced fibronectin and collagen IV deposition induced by diabetes
To investigate whether RIPK3 deficiency alleviates renal fibrogenesis, we compared WT and RIPK3 -/- mice, each with or without diabetes. The expression of the extracellular matrix (ECM) proteins fibronectin and type IV collagen were measured in the kidney cortex of mice by RT-PCR and immunohistochemical analysis. As shown in Fig. 5.3 a, elevated fibronectin mRNA expression was found in diabetic WT mice compared to non-diabetic controls (P
Consistent with the RT-PCR results, immunohistochemical analysis revealed increased staining of fibronectin and type IV collagen protein in the diabetic mice (all P

Figure 5.3 Increased fibronectin and collagen IV deposition in kidneys of diabetic mice
(a, b) Fibronectin and collagen IV mRNA expression was measured by RT-PCR in mice (WT or RIPK3-/-) +/- diabetes mellitus. Tubulin was used as reference gene. (c) Representative images of fibronectin and collagen IV stained kidney sections are from the same groups as in (a). Magnification: 20×. (d, e) Positive area percentage quantitation of fibronectin and collagen IV immunohistochemical stains are from the same groups as in (a). Statistical analysis was performed by one-way analysis of variance ANOVA followed by Tukey’s multiple comparisons test. Results are presented as mean ± S.E.M. *P
RIPK3 gene knockout decreased transforming growth factor beta (TGFβ)1 expression and myofibroblast activation
To understand the association between RIPK3 and ECM deposition we examined renal TGFβ1 mRNA expression levels in diabetic WT and RIPK3 -/- mice using RT-PCR. Induction of diabetes in WT mice led to significantly increased TGF β1 mRNA expression (Pα-smooth muscle actin (α-SMA) gene expression is commonly regarded as a specific marker of myofibroblast differentiation downstream of TGFβ1 activation. Hence α-SMA mRNA was examined using RT-PCR. The data showed that α-SMA mRNA expression levels in kidneys of WT mice with diabetes were upregulated compared to non-diabetic mice (Pdisruption of RIPK3 suppresses TGF β1 expression and myofibroblast differentiation in kidneys.
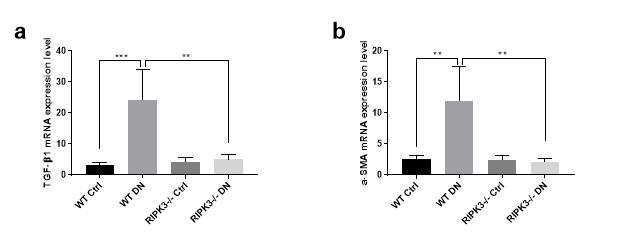
Figure 5.4Increased transforming growth factor beta (TGF β) 1 mediated myofibroblast activation in kidneys of diabetic mice.
(a, b) TGF β1 and α-smooth muscle actin (α-SMA) mRNA expression were measured by RT-PCR in mice (WT or RIPK3-/-) +/- diabetic nephropathy. Tubulin was used as the reference gene. Statistical analysis was performed by one-way analysis of variance ANOVA followed by Tukey’s multiple comparisons test. Results are presented as mean ± S.E.M. **P
RIPK3 gene knockout reduced kidney inflammatory cell infiltration
It is well known that inflammatory cell infiltration plays a crucial role in progressive diabetic kidney disease. Thus, we measured monocyte chemoattractant protein (MCP)-1 and the mRNA expression of the macrophage marker F4/80. As shown in Fig. 5.5 a, MCP-1 mRNA expression was up-regulated in kidneys of diabetic WT mice compared to non-diabetic controls. However, this upregulation was partially reversed in diabetic RIPK3 -/- mice (all P
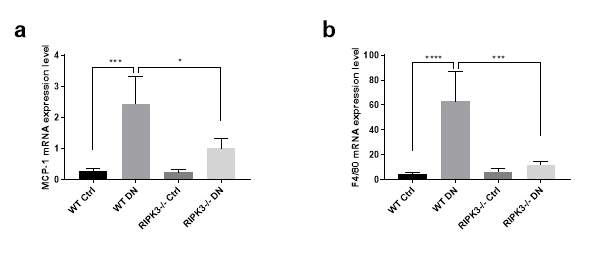
Figure 5.5 Increased inflammatory cells infiltration in the kidneys of diabetic mice.
(a, b) Monocyte chemoattractant protein (MCP)-1 and F4/80 mRNA expression were measured by RT-PCR in mice (WT or RIPK3-/-) +/- diabetes mellitus. Tubulin was used as the reference gene. Statistical analysis was performed by one-way analysis of variance ANOVA followed by Tukey’s multiple comparisons test. Results are presented as mean ± S.E.M. *P
RIPK3 gene knockout reduced TLR4 signalling activation
To understand the role of RIPK3 in renal fibrosis and to identify the mechanism whereby RIPK3 participates in TLR4 signalling, mRNA expression of TLR4 and its downstream interleukin (IL)-1β were measured using RT-PCR. As shown in Fig. 5.6 a, the expression of TLR4 was significantly up-regulated in diabetic WT mice compared to non-diabetic controls, while RIPK3 gene knockout alleviated this up-regulation. mRNA expression of TLR2 only showed a trend of inhibition by the blockade of RIPK3 (data not shown). IL-1β mRNA was elevated in kidneys of diabetic WT mice and gene ablation of RIPK3 reversed this effect (all P
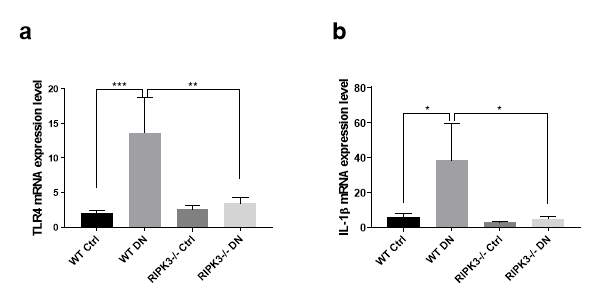
Figure 5.6 Increased toll-like receptor (TLR) 4 signalling in kidneys of diabetic mice.
(a, b) TLR4 and interleukin (IL)-1 β mRNA expression were measured by RT-PCR in mice (WT or RIPK3-/-) +/- diabetes mellitus. Tubulin was used as the reference gene. Statistical analysis was performed by one-way analysis of variance ANOVA followed by Tukey’s multiple comparisons test. Results are presented as mean ± S.E.M. *P
RIPK3 gene knockout reduced NLRP3 inflammasome formation
It is well documented that IL-1β serves as the active downstream cytokine of NLRP3 inflammasome. To examine if RIPK3 regulates NLRP3 activation in the kidneys of diabetic mice, mRNA expression of the NLRP3 inflammasome components NLRP3 and the adapter protein apoptosis‐related speck‐like protein (ASC) were examined using RT-PCR. As shown in Fig. 5.7 a, mRNA expression of NLRP3 was upregulated in kidneys of diabetic WT mice compared to non-diabetic controls (P
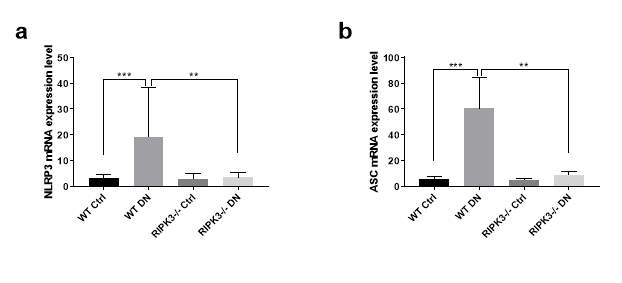
Figure 5.7 Increased pyrin domain-containing protein (NLRP) 3 formation in kidneys of diabetic mice.
(a, b) NLRP3 and adapter protein apoptosis-associated speck-like protein (ASC) mRNA expressions were measured by RT-PCR in mice (WT or RIPK3-/-) +/- diabetes mellitus. Tubulin was used as reference gene. Statistical analysis was performed by one-way analysis of variance ANOVA followed by Tukey’s multiple comparisons test. Results are presented as mean ± S.E.M. **P
Dabrafenib treatment increases survival rate in mice with established diabetic nephropathy
To study whether the RIPK3 inhibitor dabrafenib suppresses the progression of diabetic nephropathy and associated mortality, survival rates of mice after induction of diabetes were assessed over a period of 24 weeks. As shown in Fig.5.8, all control mice survived throughout the experimental period. Diabetic mice, without dabrafenib treatment, had a significantly decreased survival compared with control group (P=0.0015), reflecting a survival rate of 84.6% at day 10, decreasing to 76.9% at day 14. Further attrition occurred at the 134th, 135th day, 166th day and 167th day) resulting in a 46.2% survival at the time of culling. However, dabrafenib treatment conferred an increased survival rate at each time point compared to untreated mice reflected in a survival rate of 75% after 24 weeks of diabetes. This data demonstrates that dabrafenib confers a survival benefit in the animals with diabetic kidney disease.
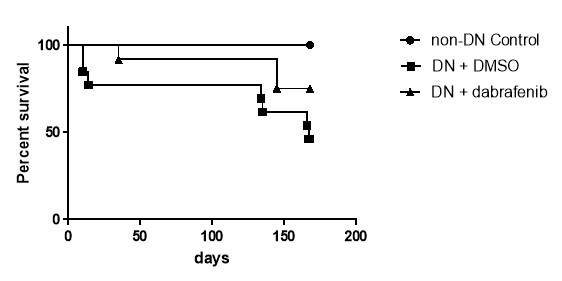
Figure 5.8 Decreased survival rate in diabetic mice
Dabrafenib treatment reduces ECM deposition in mice with established diabetic nephropathy
To determine if dabrafenib alleviates diabetes-induced renal fibrosis, we performed immunohistochemistry to measure ECM deposition in the kidney. As shown in Fig.5.9 a and b, diabetic mice had significantly higher renal cortical collagen I expression, whereas this increase was reversed by dabrafenib treatment (P
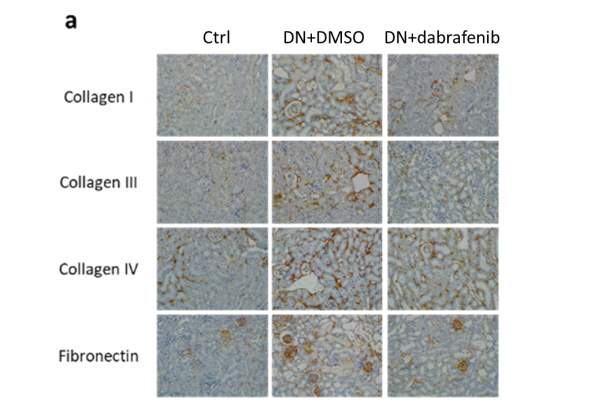
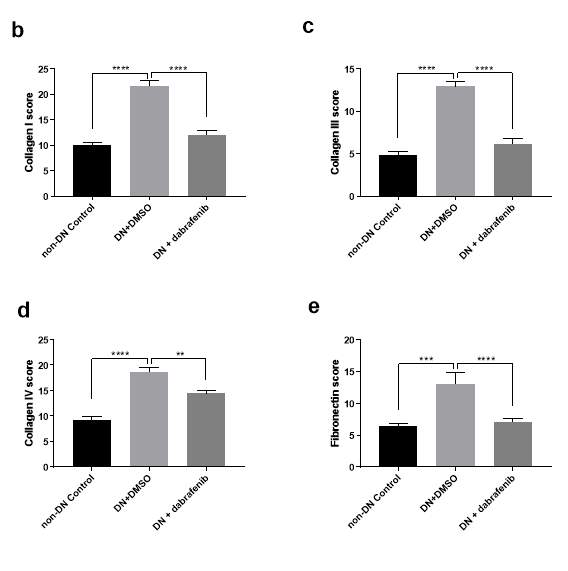
Figure 5.9 Increased extracellular matrix (ECM) deposition in kidneys of diabetic mice.
(a) Representative immunohistochemical staining images of collagen I, collagen III, collagen IV and fibronectin in the renal cortex from eNOS-/- mice (with or without treatment) +/- diabetic nephropathy. Magnification: 20×. (b-e) The quantitation of collagen I, collagen III, collagen IV and fibronectin expression in mice kidneys. Statistical analysis was performed with ANOVA followed by Tukey’s multiple comparisons test. Results are presented as mean ± S.E.M. *, P
Dabrafenib treatment decreased myofibroblast activation and inflammatory response
Given having demonstrated the upregulation of ECM and inflammasome markers in diabetic animal, we investigated whether dabrafenib reduced these markers predictive of CKD in diabetic mice. As shown in Fig 5.10 a and b, dabrafenib didn’t change the TGFβ expression but show a trend to reducing α-SMA (P=0.0643). Dabrafenib also suppressed the inflammatory response with a reduced MCP-1 (Fig. 5.10 c, P
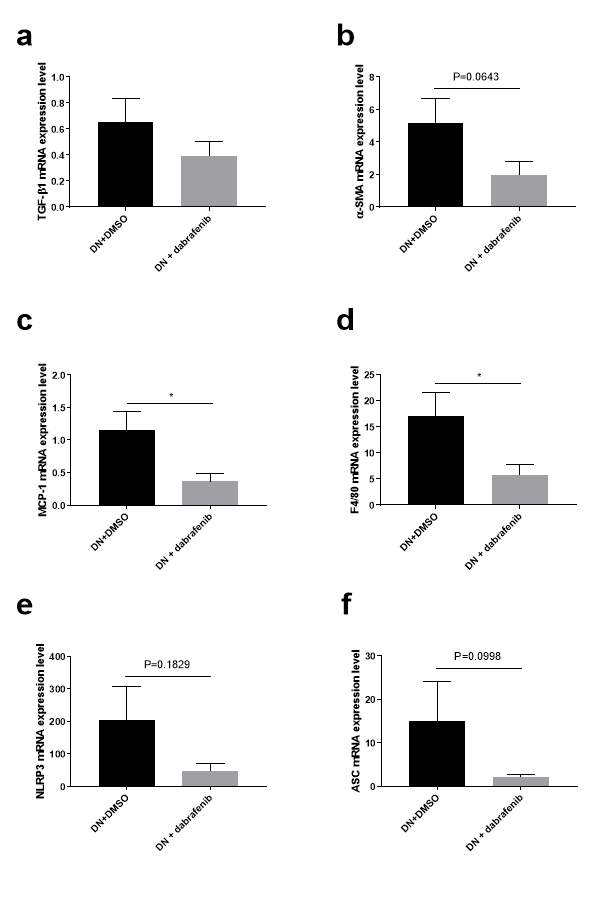
Figure 5.10 Dabrafenib reduced the inflammatory response and trended to a reduction in mRNA expression of TGFb1 and the inflammasome in kidneys of diabetic mice.
(a-f) mRNA expressions of TGFβ, α-SMA, MCP-1. F4/80, NLRP3 and ASC were measured by RT-PCR in the renal cortex from eNOS-/- diabetic mice (with or without treatment). Statistical analysis was performed using two-tailed t-tests. Results are presented as mean ± S.E.M. *, P
Dabrafenib treatment decreased myofibroblast activation in mice with established diabetic nephropathy
Given no significant difference was observed in TGF1 mRNA but trends were observed to a reduction in α-SMA mRNA expression, we measured the α-SMA protein expression within the kidney cortex to investigate if dabrafenib reduces myofibroblast activation. As shown in Fig.5.11 a and b, diabetic mice showed significantly elevated α-SMA protein expression, whereas this effect was significantly reduced in mice exposed to dabrafenib (P
To assess whether dabrafenib RIPK3 reduced TGFβ downstream Smad signalling independent of mRNA expression, we measured the p-Smad2/3 within the kidney cortex. As shown in Fig. 5.11 a and c, diabetes stimulated a significant up-regulation of p-Smad2/3 within the kidney cortex, which was decreased in dabrafenib treated mice similarly exposed to STZ (P
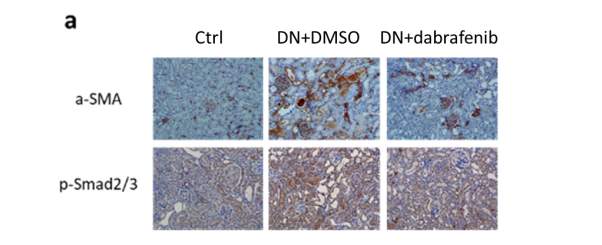
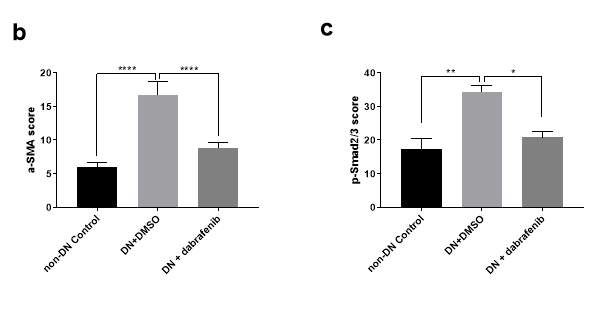
Figure 5.11 Increased ECM deposition and pSmad2/3 expression in kidneys of diabetic mice.
(a) Representative immunohistochemical staining images of α-SMA and p-Smad2/3 in the renal cortex from eNOS-/- mice (with or without treatment) +/- diabetic nephropathy. Magnification: 20×. (b-c) The quantitation of α-SMA and p-Smad2/3 expression in mice kidneys. Statistical analysis was performed with ANOVA followed by Tukey’s multiple comparisons test. Results are presented as mean ± S.E.M. *, P
Discussion
This study was undertaken to define the role of RIPK3 in regulating tubulointerstitial fibrosis in diabetic nephropathy. Significant up-regulation of RIPK3 was evident in the kidney cortex of animals and humans with diabetic kidney disease. RIPK3 deficiency downregulated TLR4 signalling, NLRP3 inflammasome activation, and TGF-β1 expression and signalling, associated with a less inflammatory cell infiltration, myofibroblast activation and less ECM deposition. The RIPK3 inhibitor dabrafenib also replicates these renoprotective effects.
Evolving data suggest that TLR4 signalling and the associated downstream cytokine IL-1β drives renal inflammation and fibrosis [10, 20-22]. Our group has previously shown that blocking TLR4 protects the kidney against tubular injury in in vivo and in vitro models of diabetic kidney disease [5, 23]. The NLRP3 inflammasome, the alternate source of IL-1β, also contributes to the progression of renal fibrosis [24]. It has been reported that NLRP3 deletion protects against renal fibrosis and attenuates inflammation in diabetic mice [25] and mice with 5/6 nephrectomy [26]. In the present study, we found genetic deletion of RIPK3 reduced TLR4 signalling as well as NLRP3 inflammasome activation, associated with reduced ECM deposition and inflammatory cell filtration. Thus, we conclude the benefit of RIPK3 inhibition on inflammatory response may arise from inhibition of TLR4 and NLRP3 activation, in addition to reduced expression and/or downstream signalling of TGF1.
In our study, TGF-β1 was reduced by RIPK3 deficiency. Although no change in expression of TGF1 was observed in diabetic animals treated with dabrafenib, decreased phosphorylation of p-Smad2/3was observed. A recent study also reported that TGF-β1 expression was not altered by RIPK3 deficiency in a 7 days unilateral ureteral obstruction (UUO) mouse mode,l which is well recognised as a fibrogenic model. In this model, RIPK3 contributes to fibrosis via regulating proliferation of fibroblasts [16]. Hence RIPK3 may contribute to fibrosis through multiple pathways, depending on the cell type and the context of the fibrotic stimuli. However, the downregulation of TGF-β1 expression and/or downstream signalling by RIPK3 inhibition may still contribute to the reduction in renal fibrosis.
It is well documented that activation of TLR4 and the NLRP3 inflammasome triggers downstream profibrotic pathways [27], including stimulation of TGF1 expression and signalling [25, 28], which serves to amplify the fibrotic response [29, 30]. Hence it is possible that inhibition of RIPK3 may directly inhibit TGFβ responses, or indirectly as a consequence of suppression of TLR4 and the NLRP3 inflammasome.
There are several potential reasons why dabrafenib was not as potent in reducing inflammasome activation as was demonstrated in the RIPK3-/- animal. It is inevitable that the animals with genetic deletion of RIPK3 have greater inhibition of RIPK3 compared with a pharmacological inhibitor. Serum assays to determine the level of RIPK3 inhibition were not undertaken throughout the study. Furthermore, DMSO was used as the vehicle for dabrafenib delivery in our study. Despite the demonstrated toxicity of DMSO [31-33], it is recognised that DMSO administration may also lead to reduced transcription of inflammatory pathways [34,35]. Low concentrations of DMSO have been found to suppress CD4+ T cell activation, function, and metabolism [36]. A study has also demonstrated that DMSO attenuates NLRP3 inflammasome activation in macrophages and LPS-induced mice [37].Hence, the magnitude of inflammation observed in the diabetic animals treated with DMSO may have been diminished. Therefore, the benefit of dabrafenib may have been attenuated.
In summary, the results from these animal studies have demonstrated that RIPK3 is an important mediator of tubulointerstitial fibrosis in diabetic kidney disease. Furthermore, we have shown that blockade of RIPK3 exerts antifibrotic effects in the kidney that may arise due to a combination of inhibition of TLR4 signalling, NLRP3 inflammasome activation and TGF1 expression and downstream signalling.
References
Li, P.K. and T.K. Ma, Global impact of nephropathies. Nephrology (Carlton), 2017. 22 Suppl 4: p. 9-13.
Kim, M.K., Treatment of diabetic kidney disease: current and future targets. Korean J Intern Med, 2017. 32(4): p. 622-630.
He, S., et al., Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A, 2011. 108(50): p. 20054-9.
Lawlor, K.E., et al., RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun, 2015. 6: p. 6282.
Ma, J., et al., TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS One, 2014. 9(5): p. e97985.
Chen, L., et al., Relaxin abrogates renal interstitial fibrosis by regulating macrophage polarization via inhibition of Toll-like receptor 4 signaling. Oncotarget, 2017. 8(13): p. 21044-21053.
Pushpakumar, S., et al., Toll-like Receptor 4 Deficiency Reduces Oxidative Stress and Macrophage Mediated Inflammation in Hypertensive Kidney. Sci Rep, 2017. 7(1): p. 6349.
Wang, S., et al., Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis, 2017. 8(7): p. e2937.
Wada, J. and H. Makino, Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol, 2016. 12(1): p. 13-26.
Yiu, W.H., M. Lin, and S.C. Tang, Toll-like receptor activation: from renal inflammation to fibrosis. Kidney Int Suppl (2011), 2014. 4(1): p. 20-25.
Spagnolo, F., P. Ghiorzo, and P. Queirolo, Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget, 2014. 5(21): p. 10206-21.
Odogwu, L., et al., FDA Approval Summary: Dabrafenib and Trametinib for the Treatment of Metastatic Non-Small Cell Lung Cancers Harboring BRAF V600E Mutations. Oncologist, 2018.
Gerarduzzi, C., et al., Prostaglandin E2-Dependent Phosphorylation of RAS Inhibition 1 (RIN1) at Ser 291 and 292 Inhibits Transforming Growth Factor-beta-Induced RAS Activation Pathway in Human Synovial Fibroblasts: Role in Cell Migration. J Cell Physiol, 2017. 232(1): p. 202-15.
Cruz, S.A., et al., Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen Res, 2018. 13(2): p. 252-256.
Li, J.X., et al., The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis, 2014. 5: p. e1278.
Imamura, M., et al., RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight, 2018. 3(3).
Yang, X.S., et al., Hypoxia-inducible factor-1 alpha is involved in RIP-induced necroptosis caused by in vitro and in vivo ischemic brain injury. Sci Rep, 2017. 7(1): p. 5818.
Huang, C., et al., Blockade of KCa3.1 ameliorates renal fibrosis through the TGF-beta1/Smad pathway in diabetic mice. Diabetes, 2013. 62(8): p. 2923-34.
Doi, S., et al., Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem, 2011. 286(10): p. 8655-65.
Souza, A.C., et al., TLR4 mutant mice are protected from renal fibrosis and chronic kidney disease progression. Physiol Rep, 2015. 3(9).
Hughes, M.M. and L.A.J. O’Neill, Metabolic regulation of NLRP3. Immunol Rev, 2018. 281(1): p. 88-98.
Zhang, M., et al., Chop deficiency prevents UUO-induced renal fibrosis by attenuating fibrotic signals originated from Hmgb1/TLR4/NFkappaB/IL-1beta signaling. Cell Death Dis, 2015. 6: p. e1847.
Mudaliar, H., et al., The role of Toll-like receptor proteins (TLR) 2 and 4 in mediating inflammation in proximal tubules. Am J Physiol Renal Physiol, 2013. 305(2): p. F143-54.
Lorenz, G., M.N. Darisipudi, and H.J. Anders, Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol Dial Transplant, 2014. 29(1): p. 41-8.
Wu, M., et al., NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol Cell Endocrinol, 2018.
Gong, W., et al., NLRP3 deletion protects against renal fibrosis and attenuates mitochondrial abnormality in mouse with 5/6 nephrectomy. Am J Physiol Renal Physiol, 2016. 310(10): p. F1081-8.
Lemos, D.R., et al., Interleukin-1beta Activates a MYC-Dependent Metabolic Switch in Kidney Stromal Cells Necessary for Progressive Tubulointerstitial Fibrosis. J Am Soc Nephrol, 2018. 29(6): p. 1690-1705.
Lin, M., et al., The TLR4 antagonist CRX-526 protects against advanced diabetic nephropathy. Kidney Int, 2013. 83(5): p. 887-900.
Otto, G., IL-1beta switches on kidney fibrosis. Nat Rev Nephrol, 2018. 14(8): p. 475.
Nee, L.E., et al., TNF-alpha and IL-1beta-mediated regulation of MMP-9 and TIMP-1 in renal proximal tubular cells. Kidney Int, 2004. 66(4): p. 1376-86.
Noel, P.R., et al., The toxicity of dimethyl sulphoxide (DMSO) for the dog, pig, rat and rabbit. Toxicology, 1975. 3(2): p. 143-69.
Galvao, J., et al., Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J, 2014. 28(3): p. 1317-30.
Yuan, C., et al., Dimethyl sulfoxide damages mitochondrial integrity and membrane potential in cultured astrocytes. PLoS One, 2014. 9(9): p. e107447.
Kilincaslan, H., et al., Protective effect of dimethyl sulfoxide on stricture formation in corrosive esophageal burns in rats. Eur J Pediatr Surg, 2014. 24(5): p. 403-9.
Man, W., et al., Dimethyl sulfoxide attenuates hydrogen peroxide-induced injury in cardiomyocytes via heme oxygenase-1. J Cell Biochem, 2014. 115(6): p. 1159-65.
Holthaus, L., et al., CD4(+) T cell activation, function, and metabolism are inhibited by low concentrations of DMSO. J Immunol Methods, 2018.
Ahn, H., et al., Dimethyl sulfoxide inhibits NLRP3 inflammasome activation. Immunobiology, 2014. 219(4): p. 315-22.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biomedical Science"
Biomedical Science focuses on how cells, organs and systems function in the human body and underpins much of modern medicine. Biomedical Science applies parts of natural and/or formal sciences to help develop advances in healthcare.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: