High Performance Liquid Chromatography
Info: 8786 words (35 pages) Dissertation
Published: 9th Dec 2019
Tagged: Chemistry
Introduction
What is HPLC
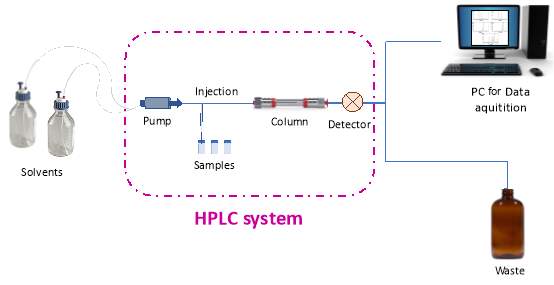
High performance liquid chromatography or high pressure liquid chromatography (HPLC) is an analytical technique used for separation, quantification or identification of compounds. HPLC has a wide range of applications such as research, food industries, pharmaceutical industries etc. An HPLC system consists of many different components. Solvents go in the system through the pump while tested samples are injected via an injector. They all pass through the column where the separation is happening and before they exit the system a detector is recording their signal.
The operating principals of this technique are based on the physical chemical properties of the compounds and the interactions between the stationary phase (SP) and the mobile phase (MP) of the system. The sample is distributing between the two phases using the liquid mobile phase as a carrier and the column content (usually silica or modified silica) as a separator.
Nowadays, various types of HPLC systems and columns are in commerce making the separation of almost any sample possible.

Figure 1: HPLC Shimadzu system
uses, how it works, system description, simadzu hplc systems
Types of HPLC
NPC, RPC, IEC, and SEC
1.2.1 Normal-Phase Chromatography (NPC) Also known as liquid-solid chromatography or adsorption chromatography, NPC is the traditional separation mode based on adsorption/desorption of the analyte onto a polar stationary phase (typically silica or alumina).3–5 Figure 1.3a shows a schematic diagram of part of a porous silica particle with silanol groups (Si-OH) residing at the surface and inside its pores. Polar analytes migrate slowly through the column due to strong interactions with the silanol groups. Figure 1.4 shows a chromatogram of four vitamin E isomers in a palm olein sample using a nonpolar mobile phase of hexane modified with a small amount of ethanol. It is believed that a surface layer of water reduces the activ- ity of the silanol groups and yields more symmetrical peaks.3 NPC is particu- larly useful for the separation of nonpolar compounds and isomers, as well as for the fractionation of complex samples by functional groups or for sample clean-up. One major disadvantage of this mode is the easy contamination of the polar surfaces by sample components. This problem is partly reduced by bonding polar functional groups such as amino- or cyano-moiety to the silanol groups. 1.2.2 Reversed-Phase Chromatography (RPC) The separation is based on analytes’ partition coefficients between a polar mobile phase and a hydrophobic (nonpolar) stationary phase.The earliest sta- tionary phases were solid particles coated with nonpolar liquids. These were quickly replaced by more permanently bonding hydrophobic groups, such as octadecyl (C18) bonded groups, on silica support. A simplified view of RPC is shown in Figure 1.3b, where polar analytes elute first while nonpolar analytes interact more strongly with the hydrophobic C18 groups that form a “liquid- like” layer around the solid silica support.This elution order of “polar first and nonpolar last” is the reverse of that observed in NPC, and thus the term “reversed-phase chromatography.” RPC typically uses a polar mobile phase such as a mixture of methanol or acetonitrile with water. The mechanism of separation is primarily attributed to solvophobic or hydrophobic intereac- tion.12,13 Figure 1.5 shows the separation of three organic components. Note that uracil, the most polar component and the most soluble compound in the mobile phase, elutes first. t-Butylbenzene elutes much later due to increased hydrophobic interaction with the stationary phase. RPC is the most popular HPLC mode and is used in more than 70% of all HPLC analyses.3,4 It is suit- able for the analysis of polar (water-soluble), medium-polarity, and some non- polar analytes. Ionic analytes can be separated using ion-suppression or ion-pairing techniques, which will be discussed in Sections 2.3.4–2.3.6 in Chapter 2. 1.2.3 Ion-Exchange Chromatography (IEC) In ion-exchange chromatography,3–5 the separation mode is based on the exchange of ionic analytes with the counter-ions of the ionic groups attached to the solid support (Figure 1.6a). Typical stationary phases are cationic exchange (sulfonate) or anionic exchange (quaternary ammonium) groups bonded to polymeric or silica materials. Mobile phases consist of buffers, often with increasing ionic strength, to force the migration of the analytes. Common applications are the analysis of ions and biological components such as amino acids, proteins/peptides, and polynucleotides. Figure 1.7 shows the separa- tion of amino acids on a sulfonated polymer column and a mobile phase of increasing sodium ion concentration and increasing pH. Since amino acids do not absorb strongly in the UV or visible region, a post-column reaction technique is used to form a color derivative to enhance detection at 550nm. Ion chromatography14 is a segment of IEC pertaining to the analysis of low concentrations of cations or anions using a high-performance ion-exhange column, often with a specialized conductivity detector. 1.2.4 Size-Exclusion Chromatography (SEC) Size-exclusion chromatography15 is a separation mode based solely on the analyte’s molecular size. Figure 1.6b shows that a large molecule is excluded from the pores and migrates quickly, whereas a small molecule can penetrate the pores and migrates more slowly down the column. It is often called gel- permeation chromatography (GPC) when used for the determination of molecular weights of organic polymers and gel-filtration chromatography (GFC) when used in the separation of water-soluble biological materials. In GPC, the column is packed with cross-linked polystyrene beads of controlled pore sizes and eluted with common mobile phases such as toluene and tetrahydrofuran. Figure 1.8 shows the separation of polystyrene standards showing an elution order of decreasing molecular size.Detection with a refractive index detector is typical. 1.2.5 Other Separation Modes Besides the four major HPLC separation modes, several others often encoun- tered in HPLC or related techniques are noted below. • Affinity chromatography9: Based on a receptor/ligand interaction in which immobilized ligands (enzymes, antigens, or hormones) on solid supports are used to isolate selected components from a mixture.The retained com- ponents can later be released in a purified state. • Chiral chromatography16: For the separation of enantiomers using a chiral-specific stationary phase. Both NPC and RPC chiral columns are available. • Hydrophilic interaction chromatography (HILIC)9: This is somewhat similar to normal phase chromatography using a polar stationary phase such as silica or ion-exchange materials but eluted with polar mobile phases of organic solvents and aqueous buffers. It is most commonly used to separate polar analytes and hydrophilic peptides. • Hydrophobic interaction chromatography4,9: Analogous to RPC except that mobile phases of low organic solvent content and high salt concen- trations are used for the separation of proteins that are easily denatured by mobile phases with high concentrations of organic solvents used in RPC. • Electrochromatography: Uses capillary electrophoresis17 (CE) equipment with a packed capillary HPLC column. The mobile phase is driven by the electromotive force from a high-voltage source as opposed to a mechan- ical pump. It is capable of very high efficiency. • Supercritical fluid chromatography (SFC)18: Uses HPLC packed columns and a mobile phase of pressurized supercritical fluids (i.e., carbon dioxide modified with a polar organic solvent). Useful for nonpolar analytes and preparative applications where purified materials can be recovered easily by evaporating the carbon dioxide. HPLC pumps and GC-type detectors are often used. • Other forms of low-pressure liquid chromatography: — Thin-layer chromatography (TLC)19 uses glass plates coated with adsorbents and capillary action as the driving force. Useful for sample screening and semi-quantitative analysis. — Paper chromatography (PC), a form of partition chromatography using paper as the stationary phase and capillary action as the driving force. — Flash chromatography, a technique for sample purification using dis- posable glass NPC columns and mobile phase driven by gas-pressure or low-pressure pumps.
Types of Equilibria in HPLC
There are several mechanisms that determine the retention time inside a column for a specific compound. Differences in retention times lead to separation between compounds. What causes the different retention times is the equilibrium of each compound between the stationary and the mobile phase. The higher the concentration of the analyte in the mobile phase, the faster it will be eluted.
The main types of equilibria are the following:
1)
Partition equilibrium. This type of equilibrium takes place when the molecules of the solute are distributed between two liquid phases. In HPLC, one liquid phase is kept immobile on a solid material, and the other is mobile (the eluent). The immobilization of the liquid to become a stationary phase in partition chromatography is achieved, for example, when the liquid is highly polar and can establish hydrogen bonds with the solid support. One such example is water on a silica surface. In this case, the mobile phase should consist of a liquid less polar than water. However, the partition equilibrium can also be applied for a nonpolar stationary phase and a more polar mobile phase. The theory of separation in partition chromatography is based on liquid/liquid extraction principles. The different molecular species, being in continuous equilibrium between the mobile and stationary phase, will be separated based on their tendency to exist in higher concentration in the mobile liquid or in the stationary liquid, in accordance with their affinity for these phases. A schematic description of the partition chromatography process is shown in Figure 1.1.3. In partition chromatography, the concept of “immobile liquid” is commonly approached in a “loose” manner. For example, a layer of adsorbed water on the surface of a silica solid support, or a layer of bonded organic chains on a silica surface (such as in the common C18 chromatographic columns), or a layer of mechanically held polymer on an inert core are all considered liquid station-ary phases for partition chromatography. The possibility of performing chromatography using two liquid phases without having one liquid phase immobilized is exploited in countercurrent chromatography. However, this subject is beyond the purpose of the present book (for details see, e.g., [2]).
Schematic description of partition equilibrium.
FIGURE 1.1.3. Schematic description of partition equilibrium.
Figure options
2)
Adsorption equilibrium. This type of equilibrium takes place when molecules are exchanged between a solid surface and a liquid mobile phase. Assuming that the stationary phase is very polar compared to the mobile phase, the polar molecules from the mobile phase are adsorbed on the solid stationary phase surface, while the less polar molecules are kept mainly in the mobile phase. Being in equilibrium between the solid and the liquid, the more polar molecules also elute from the chromatographic column, but later than the less polar compounds. A schematic description of the adsorption chromatography process is shown in Figure 1.1.4. The partition and the adsorption are utilized basically as models for describing the type of equilibrium, but a difference between the two processes is not commonly apparent from a thermodynamic point of view [3]. Also, in many instances the separation can be viewed either as a partition or as an adsorption, the differentiation being made only with the purpose of estimating differently the separation parameters, while the classification has no effect on the real process.
Schematic description of adsorption equilibrium.
FIGURE 1.1.4. Schematic description of adsorption equilibrium.
Figure options
3)
Equilibria involving ions. Equilibria between ions in solutions take place in numerous chemical reactions. For applications in HPLC, one ionic species must be immobilized, for example, by being connected through a covalent bond to a solid matrix. One example of this type of ion can be a sulfonic group connected to polystyrene. The ions in solution can be bound by ionic interactions to the immobilized counterion or may remain in solution. The equilibrium between solid phase and mobile phase, depending on the strength of the bond to the stationary phase, may provide a means for separation. A schematic description of the interactions in the ion-exchange chromatography process is shown in Figure 1.1.5.
Schematic description of ion exchange proces.
FIGURE 1.1.5. Schematic description of ion exchange proces.
Figure options
4)
Equilibria based on size exclusion. Size exclusion uses a stationary phase that consists of a porous structure in which small molecules can penetrate and spend time passing through the long channels of the solid material, while large molecules cannot penetrate the pore system of the stationary phase and are not retained. Applied in HPLC, the large molecules elute earlier, while the small molecules are retained longer. An equilibrium can be envisioned between molecules in the mobile phase and those partly trapped in the solid matrix. A schematic description of the size exclusion process is shown in Figure 1.1.6.
Schematic description of size exclusion process.
FIGURE 1.1.6. Schematic description of size exclusion process.
Figure options
5)
Affinity interactions. This type of interaction is typical for protein binding and leads to equilibria that allow very specific separations. Examples of such interactions are protein-antibody and avidin-biotin. Affinity chromatography is widely used at low pressure for protein purification.
Different types of equilibria contribute to different HPLC separation methods. Most of those methods are listed bellow:
- Reversed-phase HPLC (or RP-HPLC)
- Hydrophobic interaction chromatography (HIC)
- Nonaqueous reversed-phase chromatography (NARP)
- Ion-pair chromatography (IPC)
- Hydrophilic Interaction Liquid Chromatography (HILIC)
- Normal-phase chromatography (NPC)
- Aqueous normal-phase chromatography (ANPC or ANP)
- Ion-exchange
- Cation-exchange chromatography
- Anion-exchange chromatography
- Ion-exclusion chromatography
- Ligand-exchange
- Ion-moderated chromatography
- size-exclusion chromatography (SEC)
- Gel filtration chromatography (GFC)
- Gel permeation chromatography (GPC)
- Chiral chromatography
- Supercritical fluid chromatography (SFC)
- Affinity chromatography
In this current study Ion-exchange chromatography is being performed through strong cation exchange (SCX) columns. SCX columns are made by attaching strong cationic groups (like -SO3–) on silica particles in a column. Given the difficulty of this venture, is has been proven that more than one mechanism takes place inside the column. Along with cation exchange interactions, hydrophobic/hydrophilic, shape discrimination and aromatic selectivity interactions take place.
In cation exchange chromatography negatively charged groups are usually attached to organic carbon chains that are immobilised in silica. The separation of the compounds is achieved via ionic interactions between negative and positive charges. The retention time is analogous to the basic or acidic strength of the analyte. In this type of separation, a buffer is usually used in order to transform the analytes to their positively charged form.
Cation-exchange chromatography is a type of HPLC used for the separation of cations (inorganic or organic). In this HPLC type the retention is based on the attraction between ions in a solution and the oppo-site charged sites bound to the stationary phase. In ion-exchange chromatography (IEC or IC) the ionic species are retained on the column based on coulombic interactions. In cation-exchange chromatography the ionic compound consisting of the cationic species M+ in solution is retained by ionic groups covalently bonded to a stationary support of the type R – X−. The ion-exchange material (e.g., an organic polymer with ionic groups) is not electrically charged, and therefore the initial form of the cation exchange already has an ionically retained cation in the form R – X−C+. The separation is achieved when different molecules in solution have different acidic or basic strength. For example, for a cation-exchange material, one species (e.g., C+) that is bound to the R – X− substrate is replaced by a stronger cationic species (e.g,. M+) such that M+ is retained from the solution, while C+ passes into the mobile phase. Two different cations from solution, M1+ and M2+, can be separated based on their retention strength.
Ion-Exchange Chromatography (IEC) In ion-exchange chromatography,3–5 the separation mode is based on the exchange of ionic analytes with the counter-ions of the ionic groups attached to the solid support (Figure 1.6a). Typical stationary phases are cationic exchange (sulfonate) or anionic exchange (quaternary ammonium) groups bonded to polymeric or silica materials. Mobile phases consist of buffers, often with increasing ionic strength, to force the migration of the analytes. Common applications are the analysis of ions and biological components such as amino acids, proteins/peptides, and polynucleotides. Figure 1.7 shows the separa- tion of amino acids on a sulfonated polymer column and a mobile phase of increasing sodium ion concentration and increasing pH. Since amino acids do not absorb strongly in the UV or visible region, a post-column reaction technique is used to form a color derivative to enhance detection at 550nm. Ion chromatography14 is a segment of IEC pertaining to the analysis of low concentrations of cations or anions using a high-performance ion-exhange column, often with a specialized conductivity detector.
A Classification of HPLC Types
A variety of HPLC types have been differentiated in the literature; some of these types are similar, and others exhibit significant differences. The differentiation was based not only on various criteria such as the nature of the stationary and mobile phases and the type of interactions assumed to lead to the separation, but also on the range of concentration of specific solvents in the mobile phase (e.g., of water) and so on. This section presents a common classification of the main types of HPLC. Because different HPLC types have different characteristics and applications, it is important to understand these differences and select the appropriate HPLC type for solving a specific separation/analysis problem.
1)
Reversed-phase HPLC (or RP-HPLC) is the most common HPLC technique, and a very large number of compounds can be separated by RP-HPLC. This type of chromatography is performed on a nonpolar stationary phase with a polar mobile phase. A wide variety of nonpolar stationary phases is available, and RP-HPLC is very likely the most common type of chromatography used in practice. The stationary phase for RP-HPLC can be obtained, for example, by chemically bonding long hydrocarbon chains on a solid surface such as silica. The most common chain bound to silica is C18 (it contains 18 carbon atoms), which has a high hydrophobic character. The bonded phase hydrophobicity may vary depending on the nature of the substituent. For example, C18 bonded phase has a higher hydrophobicity than C8 bonded phases. Polymeric materials are also used as the RP-HPLC stationary phase. The mobile phase in RP-HPLC is typically a mixture of an organic solvent (CH3CN, CH3OH, isopropanol, etc.) and water, with a range of content in the organic solvent. Small amounts of buffers can also be added to the mobile phase in RP-HPLC. The interactions in RP-HPLC are considered to be the hydrophobic forces. These forces are caused by the energies resulting from the disturbance of the dipolar structure of the solvent. The so called solvophobic effect is caused by the force of “cavity-reduction” in water around the analyte and the nonpolar stationary phase when the two are interacting. The retention of the analyte on the stationary phase is dependent on the contact surface area between the nonpolar moiety of the analyte molecule and the stationary phase, both immersed in the aqueous eluent. For this reason an analyte with a larger hydrophobic surface area (and usually with a large log Kow; see rel. 1.1.1) is more retained on the stationary phase, resulting in longer retention time compared with an analyte with a smaller hydrophobic surface (and low Kow). In RP-HPLC the separation is typically considered to be based on the partition of the analyte between the stationary phase (viewed as an immobilized liquid) and the mobile phase, although some experiments can be explained by adsorption equilibrium. The exceptional utility of RP-HPLC is based on the fact that most compounds have at least some hydrophobic moiety in their structure.
2)
Ion-pair chromatography (IPC) is applied in particular to ionic or strongly polar compounds. This type of chromatography is very similar to RP-HPLC, with the difference of having a special mobile phase (ion-pair RP). In the mobile phase of ion-pair chromatography, a reagent is added, which interacts with the ions of the analytes and forms less polar compounds that can be separated based on hydrophobic interactions with the stationary phase. For example, acids that are ionized (or very polar) can be coupled with a reagent that produces ”ion pairs” amenable to separation by RP-HPLC.
3)
Hydrophobic interaction chromatography (HIC) is a type of RP-HPLC, sometimes indicated as a milder RP-HPLC, applied to the separations of proteins and other biopolymers. The technique is based on interactions between nonpolar moieties of a protein with solvent-accessible nonpolar groups (hydrophobic patches) on the surface of a hydrophilic stationary phase (e.g., hydrophobic ligands coupled on cross-linked agarose). The promotion of the hydrophobic effect by the addition of salts (such as ammonium sulfate) in the mobile phase drives the adsorption of hydrophobic areas from the protein to the hydrophobic areas on the stationary phase. The reduction of the salting out effect by decreasing the concentration of salts in solution leads to the desorption of the protein from the solid support.
4)
Nonaqueous reversed-phase chromatography (NARP) is a RP-HPLC type utilized for the separation of very hydrophobic molecules such as triglycerides. In this type of chromatography, the stationary phase is nonpolar (similar to RP), while the mobile phase, though less nonpolar than the stationary phase, is nonaqueous (usually a mixture of less polar and more polar organic solvents) and capable of dissolving the hydrophobic molecules.
5)
Hydrophilic Interaction Liquid Chromatography (HILIC) is a type of HPLC applied for polar, weakly acidic, or basic samples. In this type of HPLC the stationary phase is polar, and the mobile phase is less polar than the stationary phase. HILIC is the “reverse” of RP-HPLC. For HILIC, the polar stationary phase is typically made by chemically bonding on a solid support molecular fragments with a polar end group (diol, amino, special zwitterionic, etc.). The chromatography performed on bare silica support with free silanol (triple bond; length of mdashSi-OH) groups can also be considered as HILIC, depending on the mobile phase. The mobile phase in HILIC is typically a less polar but water-soluble solvent such as CH3OH or CH3CN, which also contains a certain proportion of water. The separation is based on the difference in polarity between the molecules. Ion-polar interactions may also play a role in separation. Viewed as having the separation equilibrium based on the interaction of a solid surface with the molecules from a liquid, HILIC is a type of adsorption chromatography. However, a (polar) bonded phase may be seen as a stationary liquid phase, and in this case HILIC is a type of partition chromatography. When the separation is done on zwitterionic phases, HILIC chromatography is sometimes indicated as ZIC (from zwitterionic chromatography).HILIC separations can also be performed on an ion-exchange stationary phase, with the mobile phase containing a high proportion of an organic solvent. This type of separation is sometimes indicated as eHILIC or ERLIC (from electrostatic repulsion hydrophilic interaction chromatography). This technique can be cationic eHILIC or anionic eHILIC, depending on the nature of the ion-exchange stationary phase. In this type of chromatography, the ionic stationary phase repels the similar ionic groups of the analyte and allows HILIC type interactions with the neutral polar molecules of the analyte.
6)
Normal-phase chromatography (NPC) is a chromatographic type that uses a polar stationary phase and a nonpolar mobile phase for the separation of polar compounds. The nonpolar mobile phases used in this type of chromatography are solvents such as hexane, CH2Cl2, and tetrahydrofuran that are not water soluble. In normal-phase chromatography, the most nonpolar compounds elute first and the most polar compounds elute last. Normal-phase chromatography does not have a major difference from HILIC. Because NPC was identified as a separate type for a much longer time than HILIC, it is common in the literature to identify HILIC as a subtype of normal-phase chromatography and not the other way around. The difference consists in the use in HILIC of a mobile phase that contains some proportion of water. A polar organic normal phase is sometimes mentioned as a type of chromatography when the nonaqueous solvent contains polar additives such as trifluoroacetic acid.
7)
Aqueous normal-phase chromatography (ANPC or ANP) is a technique performed on a special stationary phase (silica hydride), and the mobile phase covers the range including the types used in reversed-phase chromatography and those used in normal-phase chromatography. The mobile phases for ANP are based on an organic solvent (such as methanol or acetonitrile) with a certain amount of water such that the mobile phase can be both “aqueous” (water is present) and “normal” (less polar than the stationary phase). Polar solutes are most strongly retained in ANP, with retention decreasing as the amount of water in the mobile phase increases.
8)
Cation-exchange chromatography is a type of HPLC used for the separation of cations (inorganic or organic). In this HPLC type the retention is based on the attraction between ions in a solution and the oppo-site charged sites bound to the stationary phase. In ion-exchange chromatography (IEC or IC) the ionic species are retained on the column based on coulombic interactions. In cation-exchange chromatography the ionic compound consisting of the cationic species M+ in solution is retained by ionic groups covalently bonded to a stationary support of the type R – X−. The ion-exchange material (e.g., an organic polymer with ionic groups) is not electrically charged, and therefore the initial form of the cation exchange already has an ionically retained cation in the form R – X−C+. The separation is achieved when different molecules in solution have different acidic or basic strength. For example, for a cation-exchange material, one species (e.g., C+) that is bound to the R – X− substrate is replaced by a stronger cationic species (e.g,. M+) such that M+ is retained from the solution, while C+ passes into the mobile phase. Two different cations from solution, M1+ and M2+, can be separated based on their retention strength.
9)
Anion-exchange chromatography is a type of HPLC used for the separation of anions (inorganic or organic). This HPLC is similar in principle to the cation-exchange type, but the anionic species B− from solution are retained by covalently bonded ionic groups of the type R − Y+. Similarly to cation exchange stationary phases, an anion exchange is initially in the form R − Y+A−. For an anion-exchange material the anion A− previously bound is replaced on the resin by the anion B−, and two different anions B1− and B2− are separated based on their different retention strengths. The mobile phase in ion-exchange chromatography frequently con-sists of buffer solutions.
10)
Ion-exchange on amphoteric or zwitterionic phases is a type of IEC that is very similar in principle to the cation-exchange or anion-exchange IEC. The stationary phase of this type of IEC contains groups that have an amphoteric character or, in the case of zwitterionic phases, both anionic and cationic groups. The mobile phase in these types of chromatography also consists of buffer solutions.
11)
Ion-exclusion chromatography is an HPLC technique in which an ion-exchange resin is used for the separation of neutral species between them and from ionic species. In this technique, ionic compounds from the solution are rejected by the selected resin (through the so-called Donnan effect), and they are eluted as nonretained compounds. Nonionic or weakly ionic compounds penetrate the pores of the resin and are retained selectively as they partition between the liquid inside the resin and the mobile phase. (The Donnan effect or Gibbs-Donnan effect describes the distribution of ions in solution in two compartments separated by a semipermeable membrane).
12)
Ligand-exchange chromatography is a type of chromatography in which the stationary phase is a cation-exchange resin loaded with a metal ion (e.g., of a transitional metal) that is able to form coordinative bonds with the molecules from the mobile phase. The elution is done with a mobile phase able to displace the analyte from the bond with the metal, and the separation is based on the differences in the strength of the interaction (of coordinative type) of these solutes with the bonded metal ion.
13)
Immobilized metal affinity chromatography is closely related to ligand-exchange chromatography and uses a resin-containing chelating groups that can form complexes with metals such as Cu2+, Ni2+, and Zn2+. The metal ions loaded on the resin still have coordinative capability for other electron donor molecules such as proteins. The retained analytes can be eluted by destabilizing the complex with the metal, for example, by pH changes or addition of a displacing agent such as ammonia in the mobile phase.
14)
Ion-moderated chromatography is an HPLC technique similar to ligand-exchange chromatography, with the difference that the stationary phase loaded with the metal ion (e.g., Ca2+, Na+, K+, Ag+, or even H+) does not form coordinative bonds with the analyte, the interactions being based mainly on polarity.
15)
Gel filtration chromatography (GFC) is a type of size-exclusion chromatography (SEC) in which the molecules are separated based on their size (more correctly, their hydrodynamic volume). In gel filtration an aqueous (mostly aqueous) solution is used to transport the sample through the column and is applied to molecules that are soluble in water and polar solvents. Size-exclusion chromatography uses porous particles with a variety of pore sizes to separate molecules. Molecules that are smaller than the pore size of the stationary phase enter the porous particles during the separation and flow through the intricate channels of the stationary phase. Small molecules have a long path through the column and therefore a long transit time. Some very large molecules cannot enter the pores at all and elute without retention (total exclusion). Molecules of medium size enter only some larger pores and not the small ones, and are only partly retained, eluting faster than small molecules and slower than the very large ones. The separation of small molecules between themselves is not typically achieved, and the technique is utilized mainly for the separation of macromolecules and of macromolecules from small molecules. GFC is sometimes indicated as aqueous SEC.
16)
Gel permeation chromatography (GPC) is another type of size-exclusion chromatography (SEC), the only difference from gel filtration being the mobile phase, which in this case is an organic solvent. The technique is used mainly for the separation of hydrophobic macromolecules (such as solutions of certain synthetic polymers). GPC is sometimes indicated as nonaqueous SEC.
18)
Affinity chromatography is a liquid chromatographic technique typically used for protein and other bio-molecule separation and commonly indicated as bioaffinity chromatography. It can be practiced on a variety of specifically made stationary phases that allow selective retention of the analytes based on affinity interactions.
19)
Chiral chromatography on chiral stationary phases is a type of HPLC used to separate chiral compounds. Only specific applications require the separation of chiral compounds, and regular chromatography is much more common than chiral chromatography. Chiral chromatography still has numerous applications, particularly in the analysis of pharmaceutical compounds. The technique typically requires chiral stationary phases containing chiral selector groups.
20)
Chiral chromatography on achiral stationary phases is also possible for some chiral solutes by using chiral modifiers in the mobile phase, although the stationary phase is not chiral.
21)
Multimode HPLC is a type of chromatography in which the column contains by purpose more than one type of stationary phase, for example, some with bonded nonpolar groups (e.g. C18), and some with ionic groups (e.g., SO3-). This type of character can be encountered unintentionally on columns made using as a stationary phase a silica support covered with silanol groups, and also with hydrophobic groups (such as C18). In most cases, the presence of two types of interactions (e.g., polar and hydrophobic) is not desirable, but in some instances dual properties of a stationary phase can be used to the advantage of the separation.
description, mechanisms
Silica-based packing materials are most widely used in high-performance liquid chromatography (HPLC), because of their mechanical stability and a wide variety of derivatizations, as well as their relatively higher column efficiency
Strong cation exchange hplc in detail
Strong cation exchange (SCX) chromatography has been utilized as an excellent separation technique that can be combined with reversed-phase (RP) chromatography
An initial separation proceeds in a primary ion-exchange column (the particles of which shows no loss or gain of charge with varying pH) and parts of the eluent from this column are directed into a secondary column where the stationary phase is significantly less polar than the mobile phase.
Strong cation exchange groups are bound to the silica surface for a strong selectivity of positively charged compounds.
what is the protocol proposed last year
Following preliminary work, a characterisation protocol was proposed consisting of three tests. Using four basic analytes – two hydrophobic and two hydrophilic – this protocol investigates the ion exchange capacity, hydrophobic character and the synergistic effect between the two on a range of 13 different SCX phases. A number of different characterisation parameters will be used to carry out data analysis and investigate the retention mechanisms.
it was determined that SCX phases have a synergistic effect between ion exchange and hydrophobic retention mechanisms and even at high organic concentrations (70 % acetonitrile) not all other mechanisms are suppressed giving ion exchange only conditions. Also, at higher than 70 % acetonitrile, SCX columns appear to possibly be entering HILIC conditions.
From the PCA of all 13 phases, it is apparent that they are not similar with a wide spread in the score plot. From these results, it can be concluded that the characterisation protocol is discriminating with a wide variation between each parameter for all of the phases.
how do we test the robustness
a robust methodology that is affected minimally by external sources of variability through understanding how these variables impact the final quality of the method results
e identification of critical analytical method variables (CAMVs) that should be evaluated and refined through statistical DOEs.
Method validation in pharmaceutical analysis : a guide to best practice
Robustness The robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate variations in method parameters and provides an indication of its reliability during normal usage [46]. The evaluation of robustness should be considered during the development phase as such a strategy will minimize challenges that arise during inter-laboratory studies. For the determination of a method’s robustness, variables are modified within a realistic range and the quantitative influence of the variables is determined.The reliability of the analysis should be challenged with respect to deliberate changes in method variables. If the influence of the variable is within a previously specified tolerance, the variable is said to be within the method’s robustness range. If the results are influenced due to the change in analytical method variables, then those variables should be suitably controlled to a tighter range and a precautionary statement should be included in the procedure (control strategy) about that particular variable(s). 222 6 Method Design and Understanding More specifically, robustness is performed on a given test method, where each variable is deliberately modified to determine its effect on the method attributes (e.g., total % degradation products and assay level). Before evaluation of robustness testing, a set of system suitability parameters must be determined. This system suitability is a set of acceptance criteria for that particular method. Typical system suitability criteria for chromatographic methods include ranges for retention time (Rt), RRT (relative retention time), apparent efficiency (N, determined from the peak width at certain peak height, 5%, 10%, or 50% peak height being the most typical), resolution (Rs), tailing factor (Tf , at a certain peak height), asymmetry factor, and so on. Typically, these system suitability criteria and their respective ranges are initially defined during method development and further fine-tuned as a result of scouting robustness studies. During the final evaluation of robustness testing as part of methods validation, the series of system suitability requirements must be met to ensure that the validity of the analytical procedure is maintained. For chromatographic methods, robustness normally challenges both the chromatographic conditions and the sample preparation conditions. Examples of typical variations for sample preparations include extraction time, composition of extraction solvent, temperature of extraction medium, shaking time, sonication time, and so on. In the case of LC methods, the variables that can be deliberately modified are mobile phase composition, pH of the mobile phase (if applicable), ionic strength of aqueous portion of the mobile phase, concentration of mobile phase additives (chaotropic, ion-pairing), gradient slope (if applicable), initial hold time for gradient (if applicable), flow rate, column temperature, injection volume, autosampler temperature (if applicable), and wavelength. In the case of gas chromatographic (GC) methods, the variables that can be deliberately modified may include flow rate, injector temperature, detector temperature, gradient, and so on. In the case of dissolution methods, the variables that can be deliberately modified may include buffer concentration, ionic strength, pH, surfactant concentration, volume of media in vessel, speed (rpm), and so on. Generally, the following steps can be implemented during robustness evaluation of a method: 1) identification of the variables to be modified 2) defining the high and low levels for the variables 3) defining the responses (method attributes) and the acceptance criteria 4) selecting the appropriate experimental design based on the number of variables to investigate 5) execution of the robustness experiments 6) analysis of the results and if an experimental design was used, the statistical analysis of the results are determined for main effects and potential interaction effects 7) defining which method variables are critical (significant and above the noise level) 8) reassessing which responses (i.e., CMAs) are valid and refining the acceptance criteria for the responses, if appropriate 6.2 Analytical Quality by Design and Robustness Investigations 223 9) instilling the proper control strategies for the CMVs, including refining the system suitability criteria and/or adding new system suitability criteria.
Design of Experiments (DOE) A DOE can be used to provide the most efficient and statistically sound approach for evaluating multiple method variables, their main effects, and interactions in order to understand the magnitude of the impact of the variable on the responses (CMA). DOEs provide an excellent opportunity for screening a number of conditions generated from a limited number of experiments. As part of the DOE data evaluation, statistical tools are used to identify which method variables are critical as well as the appropriate ranges for these method variables to ensure the CMAs meet the desired acceptance criteria. Incorporation of DOEs into the development strategy, leads to increased knowledge/process understanding with the right amount of experiments compared to a univariate approach where less information is obtained per same unit number of experiments. In order to study the simultaneous variation of the variables on the considered responses, a multivariate approach using DOEs is recommended in robustness testing. DOEs can be used to measure the effects of analytical method variables (i.e., temperature, pH, ionic strength for chromatographic methods for assay/purity/content uniformity) on key response variable(s) (i.e., which will help dictate the proper system suitability requirements). Implementation of DOEs will elucidate the CAMVs. Then the identified variables should be controlled using a well-defined control strategy to ensure the CMAs’ criteria are met. A DOE is used to • optimize an analytical method; • minimize the total number of required experiments needed to come to reliable conclusions on confirmation of CMVs. DOE is very efficient compared to the conventional one method variable at a time (OVAT) approach. • define operational ranges for various method variables; • verify robustness for a range of the method variables; • determine the impact of the variables and the interaction of various method variables on the response variables; • help define the MODR; • provide analytical method understanding to solve potential future method challenges; • justify changes in the analytical method if required. In experimental designs for screening and/or optimization robustness, variables are intentionally set at fixed levels (high, low at a minimum). At a minimum, the range of selection of levels can be defined on basis of the precision or uncertainty (this depends on the uncertainty of the result and on the uncertainty related to instrument), and guidelines exist on how to determine this uncertainty [47, 48]. However, it is encouraged to explore relevant wider ranges to provide a larger design space. Continuous variables are variables that can be adjusted on a quantitative scale such as pH, ionic strength, temperature, and so on. Categorical variables (discontinuous variables) are variables that belong in groups or categories 226 6 Method Design and Understanding and cannot be put on a continuous scale, such as type of column, and type of detector. In the context of QbD, these are referred to as analytical noise factors. Response variables [49] (that generate numeric responses on a continuous scale) are measured and statistically analyzed to see whether or not any of the experimental variables that were changed in the DOE experiment has any impact on the responses. Examples of response variables that could be measured in chromatography experiments would include critical peak resolutions, Rt, signal-tonoise (S/N) ratio, and so on (these are also known as CMA). Classic types of experimental designs such as main effect screening designs (e.g., Plackett-Burman), factorial and fractional-factorial designs, central composites, and so on, can be utilized. Typically, during early development where there may be many variables to investigate (five or greater), a partial factorial experimental design or Plackett-Burman experimental design could be used. Later on in development, optimization DOEs can be used such as a full factorial (i.e., if a partial factorial design was used for the initial screening), central composite design, or partial factorials (if a Plackett-Burman was used for the initial screening), which are more applicable. Factorial and main effect designs will have each variable run at two or more levels. The number of levels is denoted by X and the number of variables is denoted by y, Xy. Using variables at only two levels will of course result in fewer experiments that need to be run for a given number of variables. A 23 full factorial will require 8 experiments, a 24 full factorial will require 16 runs, a 25 full factorial will require 32 runs, and so on. A complete factorial will allow for estimation of all main effects of each variable and all possible interactions between all the variables, which include all two -ay interactions, three-way, up to the k-way interactions for k variables in the design. One can use fractional factorials [50, 51] for designs (for two level factorials, they are often used for four or more variables, i.e., a half fraction factorial is denoted by 24−1 where the total number of experiments is eight as opposed to the full factorial of 16 experiments, hence a half factorial 8/16=1/2) to reduce the total number of experiments and still be able to estimate all of the main effects and some or all of the two-way and higher interactions [52–54]. An example of an experimental design is a full 23 factorial that is shown in Table 6.6. The three variables are ionic strength, %organic, and column temperature, each run at two levels resulting in a total of eight experiments. Full system suitability should be run for each of the experiments. It is good practice to add center points and/or replicate runs to an experimental design. These can serve the important purpose of estimating pure experimental error as well as providing a reference of a center between the low and high levels in a two-level factorial. Table 6.7 is an example of three added center experiments. Zero denotes the target level for the analytical method variable. They should be ideally run in a randomized order throughout the experiment. In addition, they should involve full replication of all of the setup conditions of the entire experiment, such as new buffer preparations (in case of HPLC) for each of the three experiments 6.2 Analytical Quality by Design and Robustness Investigations 227 Table 6.6 Experimental design for full factorial with three variables and two levels. Experiment Temperature Ionic strength % Organic 1 −1 −1 −1 2 −1 −1 +1 3 −1 +1 −1 4 −1 +1 +1 5 +1 −1 −1 6 +1 −1 +1 7 +1 +1 −1 8 +1 +1 +1 Table 6.7 Experiments at target level. Experiment Temperature Ionic strength % Organic 9 0 00 10 0 0 0 11 0 0 0 shown below, as these replicates can serve as an estimate of total error that occurs throughout the entire set of the experiments. In addition to factorial or fractional-factorial experiments, there are main effect screening experiments such as Plackett-Burman designs [55, 56], that can estimate main effects between two level factorials, but cannot estimate two-way interactions since they are confounded with the main effects. (Two-way interactions are moderately correlated with main effects.) A popular design is a 12-run design that can estimate a maximum of eight variables each at two levels and leave three degrees of freedom for error. In Table 6.8, is a listing of a Plackett-Burman for eight factors in 12 runs for factors X1 to X8 all at two levels (−1 and 1). Plackett-Burman experiments typically can be used when there are five or more variables to evaluate for robustness during development. Also, one can run up to eight factors in 16 runs (plus some replicate runs) and clearly get all main effects clear of the two-way interactions using a 28−4 fractional factorial. Setting up and analyzing experimental designs is possible with several good-quality DOE software packages, including Statgraphics, JMP, Design Ease, and so on [57]. There are many examples in the literature using the experimental design approach to challenge the robustness of a method [58–62]
https://www.sciencedirect.com/science/article/pii/S073170850000529X
Journal of Pharmaceutical and Biomedical Analysis
Test selectivities, retention factors, repeatability, reproducibility accuracy
parameters such as temperature, mobile phase pH, organic content, ionic strength, analyte load will be assessed
Aims & Objectives
A lot more work could be carried out on this project to further investigate the retention mechanisms and how the change in mobile phase conditions effect this for different phases. In addition to this, it would be interesting to characterise more of the same batches of all 13 phases to give an idea on their batch to batch reproducibility as this is a known issue for SCX phases.
Luna SCX (Strong Cation Exchange) provides excellent resolution and peak shape of basic, cationic compounds. However, most SCX columns show poor peak shape and bad resolution causing many chromatographers to ignore this important phase for small molecule method development, until now. Luna SCX contains a benzene sulfonic acid ligand providing ion-exchange reversed phase, and aromatic interactions. Such interactions make Luna SCX great as a first dimension of 2D LC applications as well as improved resolution for small molecules.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Chemistry"
Chemistry is a science involving the study of the elements and matter at the atomic and molecular level including their composition, structure, properties, behaviour, and how they react or combine.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: