Synthesis of Diiron Complexes with N-Heterocyclic Carbene Ligands As Hydrogenase Mimics
Info: 10117 words (40 pages) Dissertation
Published: 15th Nov 2021
Abstract
With current energy demands and an ever-dwindling supply of fossil fuel, it is important to decrease our dependency upon them. In order to decrease our dependency, it is necessary to move to an energy economy that is based on alternatives sources of energy. Hydrogen is an attractive source of energy in this respect for a number of reasons; it is carbon neutral, energy dense, and sustainable. The benefits of using hydrogen make its use as a primary energy source desirable. There is already a large amount of research along with working examples showing that hydrogen can be produced electrochemically using energy generated from solar or biofuel cells. Unfortunately, current electrochemical catalysts for hydrogen production have issues such as large overpotentials and reliability upon unsustainable, expensive platinum catalysts. There is alternative sources to hydrogen production, on promising source has been provided by nature using a type of metalloenzymes known as hydrogenases. There are three types of hydrogenases one of these is the Fe-only hydrogenases, these hydrogenases are the extremely efficient in hydrogen production using a catalyst based on the cheap metal iron. The [FeFe] and [NiFe] hydrogenase enzymes have been shown to efficiently catalyze the reduction of protons to dihydrogen. The [FeFe] hydrogenase enzymes contain an [Fe4S4] clusters that acts as an electron source for the active site of the molecule allowing hydrogen to be formed from protons.
N-heterocyclic carbene (NHC) ligands have become important in organometallic chemistry and homogeneous catalysis in the last couple of decades. NHCs are strong σ-donor ligands having little or moderate π accepting ability and are tunable both electronically and sterically. The chemistry of NHC structures using iron have not been as well researched, iron having the advantage of being relatively inexpensive and the lack of toxicity makes them more environmentally friendly. Phosphine’s have already shown promise for use in the hydrogenase active site. Phosphine’s and NHCs both donate electron density to metal centers with NHC complexes generally having greater electron density at the metal center than phosphine’s, which shows a great potential for hydrogen production.
The work contained in this thesis involves the synthesis of imidazolium based NHC ligands and their iron(II) complexes, with the objective of creating complexes for the use and function of the active sites of hydrogenases. Using NHCs as ligands has shown in previous work that you benefit from gaining the ability to modify the ligand both electronically and sterically, in addition to the stability and robustness has shown this can be used in order to stabilize the metal centers.
Table of Contents
1. Abstract
2. Abbreviations
3. Introduction and Literature Review
3.1 Hydrogen as a Fuel
3.2 N-Heterocyclic carbenes (NHCs)
3.3 NHC Applications and Previous Research
3.4 Hydrogenases
3.5 [NiFe] Hydrogenases
3.6 [Fe] Only Hydrogenases
3.7 [FeFe] Hydrogenases
3.8 Oxygen Sensitivity of Hydrogenases
4. Experimental Methods
4.1 IR Spectroscopy
5. Results and Discussion
7. Experimental
8. References
9. Appendix
Abbreviations
Ar aryl group
Et2O diethyl ether
i-Pr isopropyl
L neutral, 2 electron donor ligand
M metal
Me methyl
NHC N-heterocyclic carbene
NMR Nuclear magnetic resonance
(s) singlet
(d) doublet
(t) triplet
(m) multiplet
(br) broad
pdt propanedithiolate
Ph phenyl
Pr propyl
ppm parts per million
R generic alkyl
RT room temperature
THF tetrahydrofuran
X anionic ligand e.g. halogen
Introduction
Hydrogen as a Fuel
Fossil fuels are an ever decreasing and unsustainable source of energy and have environmental problems associated with them. This creates a need for energy alternatives in order to address global requirements. There are different energy alternatives such as hydroelectric energy, wind power, biomass and nuclear energy, these alternative sources are however limited it different ways, including access, waste disposal and output of energy.1
One suggested source to replace these forms is, hydrogen. Hydrogen is already important in industry as a chemical feedstock, the main use of hydrogen currently is in refineries for upgrading liquid fuels and in the Haber–Bosch process for ammonia synthesis. Hydrogen is projected to play a role in energy as a sustainable fuel for the future and a future hydrogen economy.2 Hydrogen as a sustainable energy solution is highly attractive as an energy source and research is being done to overcome the current issues preventing its use such as generation, transport and utilization.
From pervious research and existing technology, we already know hydrogen can be used in a hydrogen-proton exchange membrane fuel cell along with solar energy to split water. Molecular hydrogen can be combusted with oxygen to produce energy and yield water as the only byproduct making the hydrogen economy a very feasible idea. 3
2H2 + O2 ⇌ 2H2O + energy
Solar energy and hydrogen have been widely accepted as an ideal combined solution for long-term sustainable energy. The basic model for this system involves the coupling of two components, a solar H2O splitting system and a Hydrogen fuel cell system.4 Among many technical problems expected with any new technology. One critical problem is the production of hydrogen through solar energy conversion and the use of hydrogen as a fuel at cheap cost. At present, the most common material used in industries and laboratories for hydrogen technologies is platinum metal.5 Platinum has the best catalytic ability currently, however it does come with a couple of major issues. Firstly, platinum is a trace metal on earth, making it very expensive, as it is a very limited resource. Secondly, platinum is easily poisoned on exposure to CO and loses its catalytic ability for either hydrogen oxidation or proton reduction. Therefore, finding a good substitute for platinum is essential in order to further possibility of a hydrogen economy.
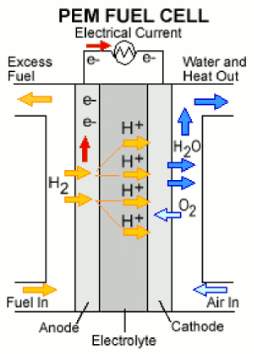
Figure 1. Basic representation of a hydrogen fuel cell. 6
An alternative to platinum for hydrogen production has already been provided by nature by means of microbial hydrogen metabolism in the form of hydrogenases. Hydrogenases have been the focus of large amounts of biological, biochemical and chemical research as biotechnologically relevant enzymes.5 In more modern research, hydrogen-activating complexes are of interest because of their significance in creating a carbon free energy economy. Hydrogenases are therefore studied in the context of understanding how to build modify and utilize catalysts wit high efficiency and ability to produce hydrogen based on hydrogenases, in hydrogen energy devices. This is a promising alternative to platinum and among the advantages already listed they use cheap, readily available elements in the functional mimic of the active site of [FeFe]-Hydrogenase.7 The major disadvantage is the oxygen sensitivity of the enzymes. 5
Research into the use of hydrogenase has already started,8-11 for both [NiFe] and [FeFe] catalysts. Fuel cells containing an Oxygen tolerant hydrogenase as anodic catalyst, with and without a membrane have been constructed to utilize the potential of hydrogenases.8 Hydrogen oxidation catalysts, which have been shown to be both selective to hydrogen and not inhibited by oxygen, make the possibility to design of hydrogen/oxygen fuel cells a real probability. Current reactions of hydrogenases with oxygen damaging the catalyst and causing degradation however, currently limit the maximum power output of enzyme based hydrogen fuel cells.
The current unsustainable but best current catalyst platinum differs in a number of ways to hydrogenases. The main way is for the homolytic cleavage of hydrogen platinum catalysts first to create a hydrogen radical which is then converted into a proton and electron.9 By contrast, Hydrogenases catalyze the heterolytic cleavage of hydrogen to create a hydride ion, which is then converted into a proton and electron. The hydrogenases by using heterolytic cleavage of Hydrogen have an advantage as this is a better approach in water.9 Since fuel cell and enzyme reactions often occur in water, heterolytic cleavage in water phase is therefore preferable in this aspect of the research into the fuel cells. Further developments into alternatives to using platinum in H2/O2 cells could come from discovery of hydrogenases with high oxygen tolerance, genetic modification of hydrogenase systems known to date, or, most likely, the synthesis of hydrogenase inspired catalysts that retain activity in the presence of oxygen as this dissertation is based on. 8, 10-11
N-Heterocyclic carbenes (NHCs)
There are two forms of carbene, triplet and singlet which are representative of the different spin states possible. The triplet carbene is the more stable of the two as it has a lower energy configuration than the singlet form.12 Triplet carbenes have two unpaired electrons, one in an sp2 orbital, one in a p orbital shown in Figure 2. Singlet carbenes have a lone pair in a nonbonding sp2 orbital, and therefore an empty p orbital also shown in Figure 2. These states are calculated by the number of electrons available for donation to a metal ion when the carbene acts as a ligand.12

Figure 2: Fischer Carbene (Triplet) and Schrock Carbene (Singlet)
N-Heterocyclic carbenes (NHCs) are deprotonated products of imidazolium salts, initial research in this area led to the synthesis of NHC complexes from electron rich olefins. 13 However the chemistry of NHCs was not investigated for a number of years as they were deemed too unstable for application in normal synthetic reactions. Arduengo first isolated free carbenes in 1991 by deprotonation of an imidazolium chloride with a strong base, causing significantly renewed interest in research into this area and the new ligands. 13
N-Heterocyclic carbenes now represent a growing class of ligands in transition-metal catalysis, due to the increased interest. N-Heterocyclic carbenes also posses a number of desirable features such as stability to air and moisture, low toxicity, strong σ-donor and poor π-acceptor properties to give high catalytic efficency.14
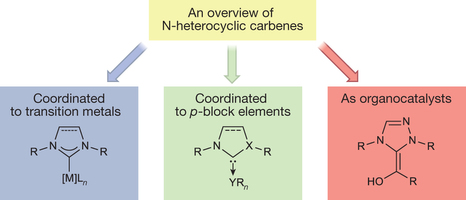
Figure 3. Major applications of NHCs. 15
Research into carbenes was influenced by the positive results gained from the binding ability of phosphines. Phosphine ligands have been used efficiently in homogeneous catalysis for long time and so much more is known about them than NHCs. Tertiary phosphines are already known to readily coordinate to a large number of transition metals and like NHCs have the ability to act as both σ-bases and π-acids, improving the stability of metals in low oxidation states.16 A phosphine-metal bond can be characterized in a similar way to an NHC, σ donation of the phosphorus lone pair to an empty p or d orbital of the metal. The π-bond originates from back-donation from the d orbital on the metal to the empty phosphine orbital. More electropositive metals will tend to form stronger σ bonds as phosphine increases the π-acidity and therefore increases the π donation from the d orbital of the metal.17 Other substituents attached to the phosphorus atoms may influence the donor or acceptor ability of the phosphine ligand.18-19
Phosphine and carbene ligands have played important roles as reactive intermediates in organometallic reactions. Since NHCs are much more stable at high temperature and pressure and can form stronger bonds to metals than phosphines, increasing the interest in such complexes. Another factor which increased interest in NHCs is that they are even more basic than any phosphine, the basicity influences the reactivity of the applied homogenous catalytic reactions they are involved in.20 However, despite strong metal–NHC bonding interactions, there are challenges associated with the chemistry of these ligands. NHC problems include that they are susceptible to loss by reductive elimination, leading to decomposition of the catalyst and therefore inactivity. This can be a big issue in some instances if the decomposition occurs before effective catalysis can take place.20
The NHC binding ability to metal centers, as ligands, has been shown to be just as good if not better than the binding ability of phosphines. Many NHC complexes of the transition metals and main group elements have been reported particularly in the palladium group.21 NHCs can bind to both hard and soft metals, which makes them a very adaptable ligand system to use and therefore very popular for research. Their binding mode was originally thought to progress through σ donation until further research showed, similar to tertiary phosphines, the occurrence of a significant degree of π back-donation.22-23 The use of these ligands in organometallic research has greatly stimulated the development of efficient catalysts based on NHCs. Many metal complexes with NHC ligands are applied as highly efficient catalysts in transformations of organic compounds. The activity and robustness of the NHC complexes has shown in many cases to surpass those of their corresponding phosphine counterparts. Combined with the stability and other benefits previously described it gave great promise that it could be used as a ligand and electron source in the hydrogenase active site.
Application of NHCs and Previous Research
Due to the strong σ-donor ability and strong metal-carbon bond of NHC ligands, they have been applied as directing ligands in numerous catalytic transformations.24 Arduengo’s discovered stable N-heterocyclic carbenes in 1991, extensive research efforts have focused on the design and application of carbenes as ligands in catalysis.13 NHCs as spectator ligands in homogeneous catalysis were reported yielding product in high yield with palladium N-heterocyclic carbene complexes. Nucleophilic N-heterocyclic complexes attracted growing interest in other areas of catalysis. Significant advances in catalytic performances have been achieved with nucleophilic carbene ligands in various catalytic reactions.
Recently, the importance of iron, highly abundant and therefore cheap, with low toxicity, has been recognized and employed in the activation and transformation of substrates.25 Fe-NHCs have been found useful also in catalytic applications for C-C bond formation, cyclization reactions and transformations as well as hydrogenase and nitrogenase active sites.26-27 Developing new Fe-NHC catalysts has great opportunity for research in this developing field and to produce useful, stable catalysts. Some, half-sandwich iron complexes of NHCs are known.28-29 Novel anionic iron(II) complexes have also been synthesized successfully, with varying success in catalysis.30-31
Iron is relatively cheap and non-toxic, hence any successful catalyst based on this metal will have a lot of potential both from an economic and environmental consideration. Using NHCs as a ligand stems from its adaptability and its ability to stabilize the Fe metal in both its high and low oxidation states. In addition the ligand has distinct stability advantages over phospines.
The main interest in the N-heterocyclic carbene complexes derives from the need for generating catalysts that will not be decomposed or deactivated. 32 By incorporating a weaker coordinating group on the same NHC ligand, catalytic activity is also possible and this presents the possibility of wider applications in industry.32 Research into the organometallic chemistry of diironthiolate complexes has resulted in observations that these structures showed the similarities to the synthetic species with the active site of the [FeFe] hydrogenase. Previous research in the active site with phosphine ligands would suggest that NHCs may improve the production and give greater control of hydrogenase active site, this enzyme catalyses the reversible reaction of 2H+ + 2e– ⇌ H2. 33-34
Sun in 2005 reported with PPh3 substitution as well as substitution with other tertiary phosphines.35 Pdt-type catalysts with tertiary phosphine and phosphite substitutions, such as PPh3, increase electron richness at iron and become more protophilic.35 Catalysts with good donor ligands, increased electron richness in the Fe-Fe bond, increasing basicity. Phosphines were found to be good ligands because they have electronic characteristics similar to CN- ligands, while the steric and electronic properties can be modified, and there are no complications involving protonation of the CN- ligand.35
Gloaguen, Talarmin and coworkers studied NHC ligands as cyanide mimics in pdt-type catalysts.34 Earlier studies had shown that replacing a CO ligand with better electron-donating groups induces protonation at the Fe-Fe center.34 The IR for the NHC complexes showed three (CO) bands which showed a shift to lower frequencies than those of the bis-phosphine complex [Fe2(pdt)(CO)4(PMe3)2]. These bands were (ν(CO) 1979, 1940, 1898 cm-1).34 The IR data obtained in the study confirmed the expected greater electron donating ability of the NHC ligand relative to that of the trimethylphosphine. 34 These results opened up the possibility to use NHC groups as a surrogate for the cyanide ligand in the same way phosphines were already being utilized.34 The evidence that NHC ligands also often exhibit greater electron donating abilities than phosphines created an interest, this interest was in the possibility for higher rates of hydrogen production and is the reason they are of interest in this project for hydrogenase active site mimics.
Previous research as already mentioned, has shown good electron-donating phosphine substituted diiron derivatives as being of particular interest in the development of catalysts for Hydrogen electrolysis. The phosphine ligands enhance the electron density. When compared with the nondirectional s-type lone pair in phosphines,, NHCs form stronger σ-bonds to a wide variety of TMs. 18 This stronger donating ability of NHCs versus phosphines can be reflected by larger proton affinities found for carbenes also do not participate in π backbonding interactions with TMs as readily as phosphones. As a result, a given metal will be more electron rich as a NHC-metal complex compared to the corresponding phosphine complex, which has major implications in the rates of a variety of catalytic processes.
Hydrogenases
Hydrogenases are redox metalloenzymes occurring in anaerobic bacteria, anaerobic protists, and mitochondrial-containing eukayotes. There are three different types of hydrogenase enzymes that have been discovered and classified based on the metal atoms found at their active sites. The three hydrogenase enzymes are, the [FeFe],[NiFe], and [Fe] only-hydrogenases.36 All of the [FeFe] and [NiFe] hydrogenases can reversibly convert protons and electrons to H2, and then H2 back into protons and electrons depending upon the redox environment of the enzyme. 37
Of the three types of hydrogenase enzymes the [NiFe]-hydrogenases and [FeFe]-hydrogenases have a binuclear active site while [Fe]-hydrogenases contain a single Fe atom.38 In every enzyme, each Fe atom is coordinated to at least one carbonyl ligand.38 Most metal centers will also coordinate to one or more cyanide ligands as well.38 The hydrogenases with a binuclear center also have iron-sulfur clusters that shuttle electrons to and from the active site.38 The [FeFe]-hydrogenase is the most active of these enzymes at producing H2catalysing the reaction.38
H2 ⇌ 2H+ + 2e–
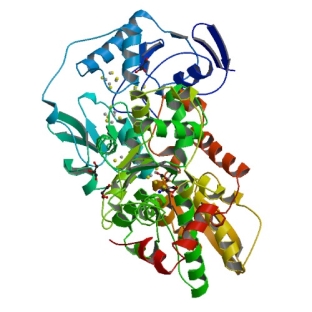
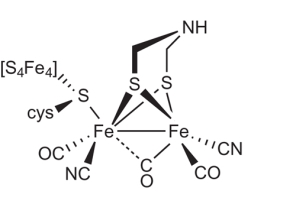
Figure 4 Angstrom crystal structure of [FeFe] hydrogenase.39
The active site shown in Figure 4 can reduce protons to Hydrogen at a high rate of 1000 molecules per second per active site with small overpotentials compared to current alternative catalysts.7 This is one of the reasons the [FeFe]-hydrogenase active site has been the focus of previous research including the work in this project.
[NiFe] hydrogenases
[NiFe] hydrogenases are found in organisms that are capable of living in very diverse environmental conditions, under light or under anoxic dark conditions.40 The structures and catalytic properties of [NiFe] hydrogenases are exactly those needed to meet the physiological requirements of the organism they are used in. The active sites of [NiFe] hydrogenases have been shown by IR spectroscopic studies to comprise of a dinuclear nickel and iron center with CO and CN– ligands. A large number of [NiFe] hydrogenases react with oxygen under electron deprived conditions to form a low energy state that is slow to reactivate under reductive conditions.41 This low energy state is often described as the unready state. In the unready state, a peroxide ligand is thought to bind in the bridging position between the Ni and Fe atoms.41 Under oxidizing potentials, [NiFe] hydrogenases experience anaerobic inactivation to form what is known as the ready state, this state is formed by reaction with oxygen under electron rich conditions.41 In the ready state, a hydroxide ligand is bound in the bridging position.41 The active site is coordinated to the protein by four cysteine residues.42 Crystallography studies have shown that three iron-sulfur clusters form a ‘tunnel’ to the center of the enzyme where the active site is located. This ‘tunnel’ consists of an [3Fe-4S] cluster between two [4Fe-4S] clusters.43. The low-spin Fe(II) ion is redox inactive throughout catalysis, whereas the Ni changes its oxidation state. In the inactive oxidized form, a third bridging ligand (OH) is found between Ni and Fe atoms.44 Once the enzyme is activated by hydrogen, the bridging position is vacant or a hydride will occupy the site. By acting in sequence, the iron- sulfur clusters provide an electron-transfer relay system, allowing electron movement through the protein matrix.44 As well as this gas channels provide an easy route for hydrogen to travel between the active site and the exterior of the protein where it can be utilised.44
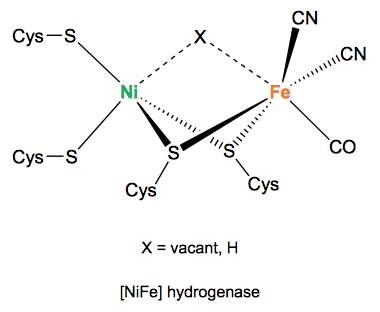
Figure 5. Active site of [NiFe] hydrogenase 45
[Fe] only hydrogenases
Unlike the other two types, [Fe]-only hydrogenases are found only in some hydrogenotrophic methanogenic archaea, a type of microorganism that produces methane in anoxic conditions.46 They also feature an entirely different enzymatic mechanism in terms of electrons transfer to the active site.46 In the alternative [NiFe] and [FeFe] hydrogenases, electrons pass through the complex via a series of metal-organic clusters over a relatively a long distance. During this process the active site structures remain unchanged during the whole process.46 However, in [Fe] only hydrogenases electrons are delivered directly to the active site over a far shorter distance. [Fe] only hydrogenase do not contain any iron-sulfur clusters unlike the other hydrogenases and requires only one metal atom for function.46 The Fe center in [Fe] only hydrogenase, however, has a similar electronic structure and is ligated by similar ligands as the distal Fe centers in [FeFe] and [FeNi] hydrogenases.46 The composition and structure of the Fe-containing active site have been revealed using a wide range of spectroscopic and crystallographic methods.46 There is still some uncertainty with the model, however currently model suggests that the Fe ion is coordinated to two cis-CO ligands, a cysteine sulfur atom, a bidentate pyridone molecule through its nitrogen and acyl carbon atoms, and what is an unidentified ligand (Figure 6).46 Methenyl-H4MPT+, a cofactor, directly accepts the hydride from Hydrogen in the process. [Fe]-only hydrogenase is also known as hydrogen forming methylenetetrahydromethanopterin (methylene-H4MPT) dehydrogenase, due to its function is the reversible reduction of methenyl-H4MPT+ to methylene-H4MPT and a proton (H+).47-48 The hydrogenation of a methenyl-H4MPT+ occurs instead of hydrogen production, which is different to the other two types of hydrogenases.47, 49 Further research is still required to obtain the exact mechanism of the catalysis which is still being studied. The most recent studies suggest that the molecular hydrogen is first heterolytically cleaved by Fe(II), followed by transfer of hydride to the carbocation of the acceptor, the reaction for hydride transfer reaction catalyzed by [Fe]-hydrogenase can be seen in Scheme 1..49
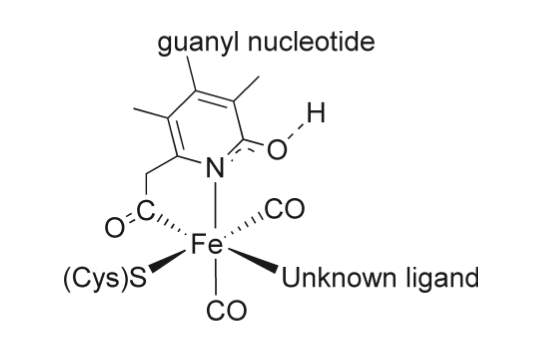
Figure 6. Proposed active site of [Fe]-hydrogenase. 46
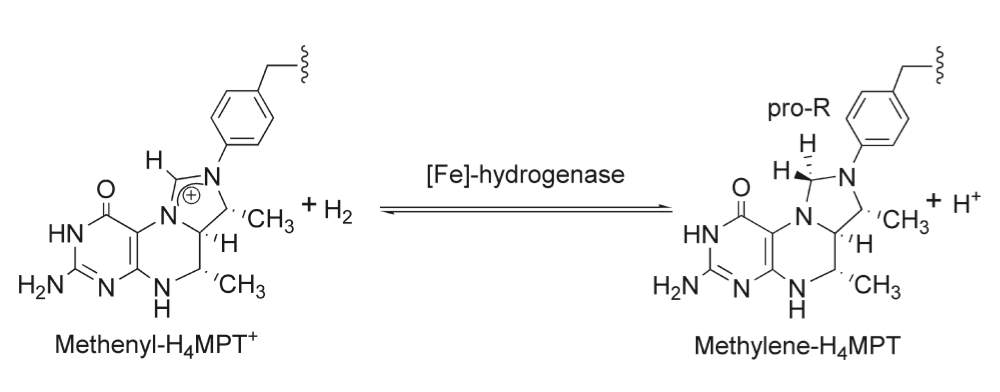
Scheme 1. Hydride transfer reaction catalyzed by [Fe]-hydrogenase. 46
The [FeFe] Hydrogenases
The structure, chemical reactivity, and mechanism of the [FeFe] hydrogenase active site are of great interest in relation to the production of energy as hydrogen gas. The [FeFe] hydrogenase active site does this by reduction of protons and to the release of energy by H–H bond cleavage in a fuel cell.50 Breakthroughs have been made in revealing the structure of the [FeFe] hydrogenase active site using high quality X-ray structures in late 1998. 50
The X-ray crystal structures have shown that the active site of [FeFe]-Hydrogenase is composed of a 4Fe4S cluster bound by a cysteine residue to the iron-sulfur sub-cluster and the two Fe atoms are linked by two sulfur bridges in which the two Fe atoms are coordinated by three CO ligands and two CN−.51 Subsequent theoretical and experimental studies give strong support to the suggestion that the two S atoms are connected by a secondary amine-containing linkage, CH2NHCH2.52 There is also biophysical evidence from experimental results calculations which have revealed that the amine cofactor acts as a shuttle for protons transferring to and from the distal Fe. In this way the 4Fe4S cluster functions as an electron transfer relay to complete the redox process at the diiron core.52 The proton-coupled electron transfer process provides a low energy pathway for H–H bond cleavage and formation at the diiron dithiolate core. The low energy pathway for electron transfer then enables [FeFe] hydrogenases to act as highly active proton reduction and hydrogen oxidation catalysts. 52
The current understanding of the biological cycle for hydrogen evolution by [FeFe] hydrogenase based upon chemical evidence and fourier transform infrared spectroscopy is represented in Scheme 2. 51
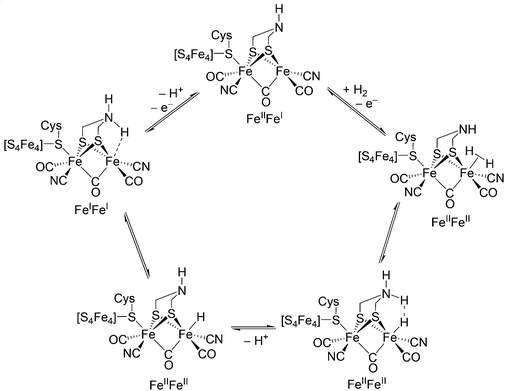
Scheme 2. Catalytic cycle for [FeFe]-hydrogenase.52 This Scheme includes assignments of oxidation states and structures for the subsite representative of a consensus view of the catalytic cycle. The scheme includes proposed intermediates although not all have yet been observed. 51-54
Oxygen Sensitivity of Hydrogenases
The [FeFe] hydrogenase catalytic site H cluster is a complex iron sulfur cofactor that is sensitive to oxygen. The oxygen sensitivity is an important obstacle to overcome in order to use hydrogen as an energy source in water-splitting, oxygenic systems. Oxygen reacts directly with the H cluster, which results in rapid enzyme inactivation and eventual degradation, not coveted properties for a catalyst in fuel cells.55 The inactivation of [FeFe] hydrogenase by O2 is defined by steps that involve the diffusion of gases into close proximity of the catalytic site, followed by redox and chemical reactions of Oxygen with the H cluster. 55
Experimental Methods
In this work, the synthetic compounds were generally characterized by the standard techniques such as NMR (Nuclear Magnetic Resonance), IR (Infrared Spectroscopy) and elemental analysis.
IR Spectroscopy
An IR spectrum arises from the transition of energy between different vibrational modes of a molecule. In order for a vibrational mode to be IR active it must give a change in dipole moment during the vibration. Polar groups such as carbonyl and cyanide exhibit strong absorptions producing high intensity stretching bands in the IR spectrum.56 The vibrational frequencies for stretching bonds in molecules are related to the strength of the chemical bonds and the masses of the atoms. Carbonyls in this research act as reference ligands due to their sensitivity to the electron density at the metal center. This sensitivity includes changes in stretching frequencies with more of less metal carbonyl π back-donation. The lowest unoccupied molecular orbital of CO is an antibonding orbital and is low enough in energy to function as an acceptor orbital when interacting with filled d-orbitals on metals.25, 56-57 This is the reason why coordination to electron rich metal centers shifts the CO stretching frequency to lower energy as the C-O bond weakens due to the d* backbonding. An increased electron density around the two iron centers by the addition of electron-rich, strong -donor groups creates greater back-bonding to carbonyls weakens the CO bond and creates stretching frequencies at lower energies. 51
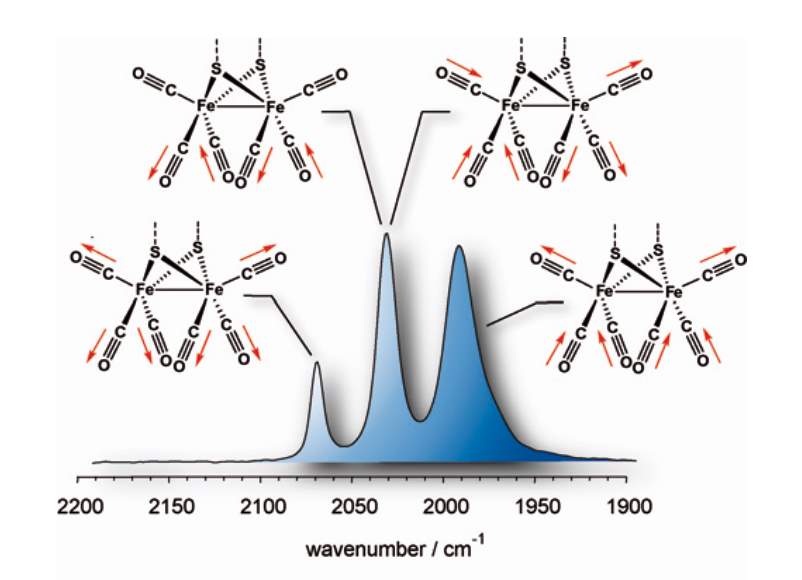
Figure 7 The assignments (most prominent showed) of the CO stretching frequencies of the IR spectra for [Fe2( -S2R)(CO)6] complexes. The IR spectrum of [Fe2(pdt)(CO)6] is shown as an example. 56
Results and Discussion
Diirion propanedithiolate hexacarbonyl synthesis
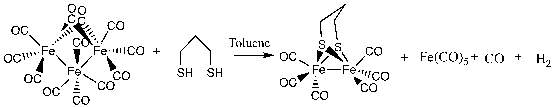
The addition of Fe3(CO)12 and 1,3-propanedithiol produced [Fe2(pdt)(CO)6] in a relatively high yield of 83.2 % in a one pot synthesis. IR confirmed the product showed the expected three CO peaks between 2100 and 2000 58. The eluent of hexane/EtOAc (80/20) for the chromatography of the products was chosen in order to elute the product was chosen to create a separation of complexes, the expected product was eluted first and the unreacted starting material remained in the column and was not collected.
Synthesis and characterisation of imidazolium salts
These reactions require Schlenk-line techniques due to their air/moisture sensitivity require Schlenk-line techniques due to their sensitivity and were also performed in an inert atmosphere. 1,3-dimethylimidazolium iodide was synthesized by addition of iodomethane to methylimidazole as the sole product of the reaction in a high yield of 90%. The reaction was a simple straightforward reaction and has been synthesized in higher quantities with a relatively similar yield. The 1HNMR showed all the expected peaks of imidazolium iodide and the one pot synthesis makes this convenient and easy to make.
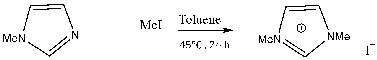
1,3-Bis(2,6-diisopropylphenyl)-4,5-dihydroimidazolium tetrafluoroborate was synthesized via N,N’-bis(2,6-diisopropylphenyl)ethylenediimine followed by cyclisation with chloromethyl ethylether which yielded both the imidazolium center and the chloride counter ion. The preparation of the tetrafluoroborate rather than keeping the chloride product was due to tetrafluoroborates tending to be less hydroscopic and easier to crystalize than chlorides.
1,3-bis(2,6-diisopropylphenyl)imidazolium chloride was obtained in two steps starting with the widely available, acyclic reagents. The condensation of glyoxal with two equivalents of 2,6-diisopropylaniline afforded the corresponding diazabutadiene. This intermediate was then cyclized into the final product using paraformaldehyde in toluene as the donor reagent and anhydrous HCl in dioxane as the source of the chloride counterion. The simple presence of aqueous HBF4 then afforded straightforward access to the tetrafluoroborate, 1,3-Bis(2,6-diisopropylphenyl)-4,5-dihydroimidazolium tetrafluoroborate. To achieve isolation of this salt it was necessary to first isolate the intermediate prior to cyclization to achieve a more efficient overall process. 59
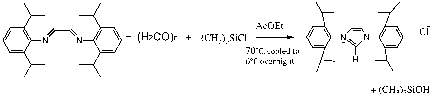
The salts were characterized at each stage of their synthesis. N,N’-bis(2,6-diisopropylphenyl)ethylenediimine showed the expected peaks as well as an amine peak accounting for unreacted 2,6-diisopropylaniline. A strong base is the most frequently used method for the generation of a free carbene based on the deprotonation of an imidazolium salt at the 2-position.
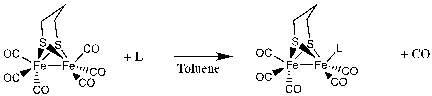
Synthesis and characterization of [Fe2(μ-S(CH2)3S)(CO)5(LMe)]
This synthesis was performed using an oxygen free environment and moisture free due to the sensitivity of free carbenes. [Fe2(μ-S(CH2)3S)(CO)5(LMe)] was prepared by adding the pdt precursor, Fe2(pdt)(CO)6], to THF in the presence of 1,3-dimethylimidazolin-2-ylidene, after reacting 1,3-dimethylimidazolium iodide with potassium tertbutoxide to produce a yellow solution as a result of the free carbene being formed. 34 1,3-dimethylimidazolium iodide was left overnight for the removal of the water to eliminate potential errors in the reaction due to the moisture sensitivity of the free carbene, this was done using Dean-Stark apparatus. The product was then filtered and chromatographed in order to separate the products. This produced one orange band and one red band, which on analysis by IR showed unreacted [Fe2(pdt)(CO)6] and not the expected shifted IR bands showing the desired reaction had not taken place.
Synthesis and characterization of [Fe2(μ-S(CH2)3S)(CO)5(L)]
This synthesis was performed using an oxygen free environment and moisture free due to the sensitivity of free carbenes. [Fe2(μ-S(CH2)3S)(CO)5(L)] was prepared by adding the pdt precursor, Fe2(pdt)(CO)6], to Toluene in the presence of 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride, after reacting 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride with potassium tertbutoxide to produce a yellow solution as a result of the free carbene being formed. 34 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride was left overnight prior to the reaction under vacuum for the removal of the water to eliminate potential errors in the reaction due to the moisture sensitivity of the free carbene. The product was then filtered and chromatographed in order to separate the products. This produced one red band and one puple band, which on analysis by IR showed unreacted [Fe2(pdt)(CO)6] in the first band and a the expected product in the second purple band eluted.
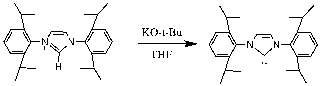
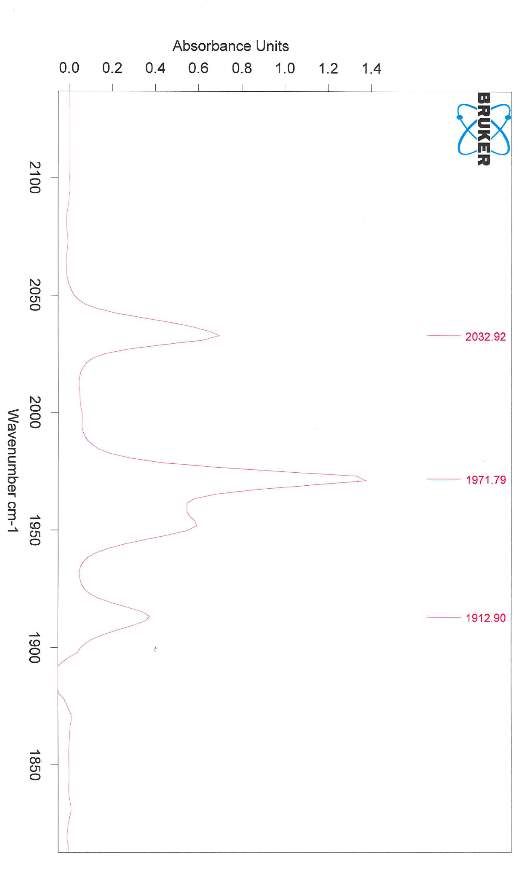
The free NHC ligand, generated by treatment of the corresponding imidazolium salt with the strong base KtBuO, was difficult to isolate and thus was kept in a THF solution at -5 °C prior to reacting with the metal complex. The IR spectra of Fe2(μ-S(CH2)3S)(CO)5(L)] displayed patterns in the carbonyl region very similar to that of [Fe2(pdt)(CO)6] but with a lower wavenumbers as expected from previous research34 suggesting that the new compound was the monosubstituted NHC metal complex desired. Shift in IR to 2033 cm-1 , 1972 cm-1 and 1913 cm-1 shows the attachment of the carbene ligand to the diiron complex. The decrease in Wavenumber of the CO stretches shows an increase in back-bonding to the metal as expected with an attached carbene ligand. The bonding between M-C strength increases, and bonding between C-O decreases because of this back-bonding and creating the shift. Unfortunately, further analysis was not possible, as the complex appeared to decompose. This was unexpected as similar phosphine based hydrogenases did not undergo the same decomposition problem. The CNMR for the complex is attached in the appendix despite decomposition having already occurred. Protonation was still attempted however confirmed that the complex had degraded as originally thought.
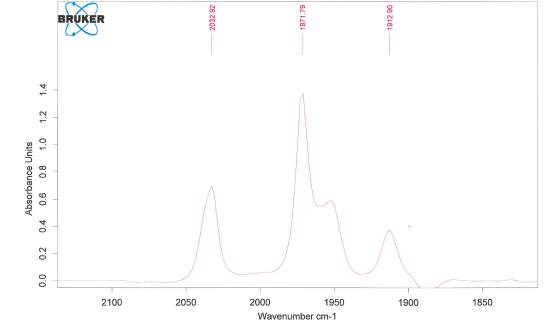
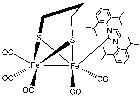
Figure 9. IR shift for the compound [Fe2(μ-S(CH2)3S)(CO)5(L)] where L is the ligand shown attached above.
Conclusion and Future Research
The original aim of this work was the preparation and characterization of [FeFe]- Hydrogenase active site inspired pdt-type catalysts with NHC ligands, then to use this ligands to determine the rate of hydrogen production possible with different NHC complexes. Using spectroscopy and electrochemistry, it was hoped insight could be gained into how to improve the catalyst to optimize the catalytic cycle. Unfortunately a number of problems encountered during this project prevented much of the intended complexes to be synthesized. The catalyst that was synthesized shown by evidence of IR shift in this dissertation also degraded before elemental analysis was received. Future directions for this type of research include obtaining further analysis and electrochemistry on the protonated complex. On preparation of NHC containing complexes it is important to ensure purification of starting materials as well as the absence of oxygen and moisture during preparation which was believed to be the hindrance in attaching the ligand to the diiron dithiolate complex. Design, synthesis, and full characterization and evaluation of the catalytic activity of the catalysts could then be performed. This could then help model and explain the possible steric and electronic changes necessary to provide the best possible complex to catalytically be inexpensive and robust hydrogen-producing catalyst.
Synthesis
All reactions involving free carbenes were performed using standard Schlenk techniques under either a dry Argon or Nitrogen atmosphere. Solvents such as n-hexane, toluene, tetrohydrafuran, diethylether were dried using standard techniques. All starting chemicals used were received from Sigma Aldrich or Alfa Aesar.
Diirion propanedithiolate hexacarbonyl synthesis
Toluene (50 ml) was added to Fe3(CO)12 (5 g, 9.93 mmol) producing a green solution. 1,3-propanedithiol (1.1 mL, 10.98 mmol)was added to this solution. The green solution was heated at reflux for 2 h, turning red after 15 min. The solution. Solvent was removed by vacuum and the crude product was subjected to silica gel chromatography using hexane/EtOAc (80/20). The solvent was evapoirated producing a red crystalline solid. (4.8 g 12.4 mmol 83.2 %) IR: (EtOAc) 2073 cm-1 (CO), 2033 cm-1 (CO), 1998 cm-1 (CO).
1,3-dimethylimidazolium iodide
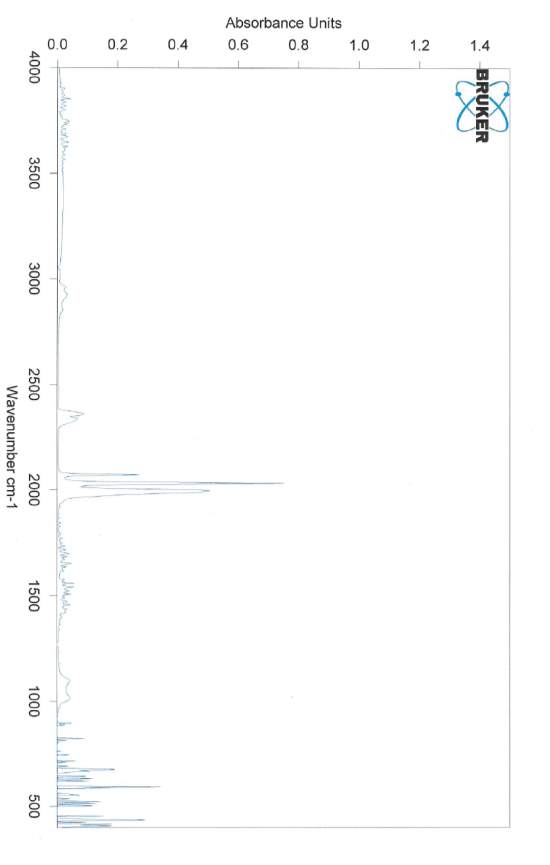
to a solution of toluene (50 ml) 1-methylimidazole (1.50 ml, 18.8 mmol) and iodomethane (1.4 ml, 22.5 mmol) were added to the solution respectively whilst stirring . The reagents were stirred at 45 °C for 24 h. After being left overnight a yellow formed. Approximately half of the toluene was decanted from the flask and the remaining solvent was evaporated and product dried. Slightly yellow crystals were obtained (3.8 g, 17 mmol 90 % yield) 1HNMR (CDCl3): δ = 10.03 (s, 1H , CH exchangeable) 7.41 (s, 2H, CH), 6.10 (s, 6H, CH3).
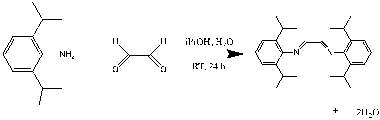
Synthesis of N,N’-bis(2,6-diisopropylphenyl)ethylenediimine
2,6-Diisopropylaniline (6.341 g , 35.7 mmol) and isopropanol (17 mL) was added to a 250 ml round bottom flask. In a 100 mL Erlenmeyer flask, glyoxal (6 ml, 17.3 mmol,) was added to water (6 mL) and isopropanol (6 mL). This solution was added to solution of 2,6-diisopropylaniline producing a yellow-brown solution, which was stirred for 20 h at room temperature. A yellow precipitate formed over the 20 h period. The yellow suspension was then filtered and the precipitate rinsed twice with water (2 ×10 mL) and dried under vacuum leaving a yellow solid (1.7 g, 4.46 mmol 25 %). 1HNMR (CDCl3): δ = 8.10 (s, 2H, CH=N), 7.16 (m, 6H, CHaromatic), 2.95 (sept,4H,CH(CH3)2), 1.28 (d,2H,NH2), (C1.22 (d, 24H, CH(CH3)2)
Synthesis of 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride
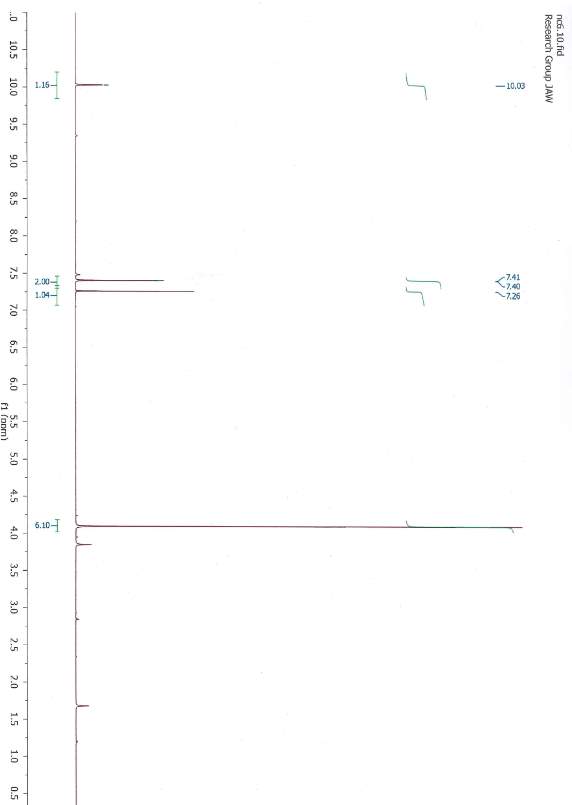
A solution of Ethyl acetate (35 mL) was heated to 70 °C. N,N’-bis(2,6-diisopropylphenyl)ethylenediimine (1.7 g, 4.51 mmol) and paraformaldehyde (0.3 g, 11.7 mmol) producing a yellow solution. Chlorotrimethylsilane (0.5 g, 0.6 mL, 4.6 mmol) in ethyl acetate (0.5 mL) was added dropwise to the solution over 45 min with stirring. The resulting solution was stirred for a further 2 h at 70 °C. The solution changed to black-brown and the mixture as it was cooled to room temperature before stored for 15 h at 5 °C. A visible white precipitate could be seen the following day and the cold suspension was filtered through a Büchner funnel. The remaining white precipitate was rinsed twice with ethyl acetate (2 ×10 mL) and once with diethylether (10 mL). The precipitate was dried leaving a white crystalline powder (1.2 g, 2.8 mmol 63 % yield). 1HNMR (CDCl3): δ = 10.09 (s, 1H, CH), 8.14 (s, 2H, CH), 7.56 (t, 2H, p-CH), 7.34 (d, 4H, m-CH), 2.44 (sept, 4H, CH(CH3)2), 1.28 (d, 12H, CH3(CH2)2), 1.24 (d, 12H, CH3(CH2)2).
Synthesis of 1,3-Bis(2,6-diisopropylphenyl)-4,5-dihydroimidazolium tetrafluoroborate
1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (1.2 g , 2.8 mmol) in 60 ml H2O. NH4BF4 (6 ml, 1 M, 6 mmol) was added forming a white precipitate instantaneously. Filtered and dissolved in CH2Cl2 in order to remove from flask and evaporated solvent. (0.52 g, 1.08 mmol 37%).
Synthesis of [Fe2(μ-S(CH2)3S)(CO)5(LMe)]
Potassium tert-butoxide (1.15 g, 1.0 mmol) and THF (50 mL) were added to 1,3-dimethylimidazolium iodide (2.14 g 1.1 mmol) producing a light yellow solution and stirred for 3 h. The reaction mixture was filtered through celite and eluted with THF. [Fe2(μ S(CH2)3S)(CO)6] (1 g, 2.59 mmol) was dissolved in the resulting solution and stirred until the starting material disappeared. The resulting deep red solution was filtered through Celitie under argon. The solvent was removed in vacuo. The crude solid obtained was chromatographed on silica gel. An orange band was eluted with a dichloromethane/hexane (20/80) mixture. A second red band eluted with dichloromethane/hexane (80/20).
Synthesis of [Fe2(μ-S(CH2)3S)(CO)5(L)]
Potassium tert-butoxide (1.15 g, 1.0 mmol) and THF (50 mL) were added to 1,3-dimethylimidazolium iodide (2.14 g 1.1 mmol) producing a light yellow solution and stirred for 3 h. Solution was stored at -5°C for 15 h. The reaction mixture was then filtered through celite and eluted with THF. The solvent was then evaporated under vacuum and yellow oil extracted in toluene (2 × 80 ml). The solution was reduced to ≈ 70 ml. [Fe2(μ S(CH2)3S)(CO)6] (1 g, 2.59 mmol) was dissolved in the resulting solution and stirred for 20 h. The resulting deep red solution was filtered through Celitie under argon. The solvent was removed in vacuo. The crude solid obtained was chromatographed on silica gel. A red band was eluted with a dichloromethane/hexane (20/80) mixture. A second purple band eluted with dichloromethane/hexane (80/20). (0.12 g, 0.16 mmol, 6.2 % yield) IR: (EtOAc) 2033 cm-1 (CO), 1972 cm-1 (CO), 1913 cm-1 (CO).
References
1. Loáiciga, H. A., Challenges to Phasing out Fossil Fuels as the Major Source of the World’s Energy. Energy & Environment 2011, 22 (6), 659-679.
2. Smith, P. R.; Bingham, A. S.; Swartz, J. R., Generation of hydrogen from NADPH using an [FeFe] hydrogenase. International Journal of Hydrogen Energy 2012, 37 (3), 2977-2983.
3. Hong, G.; Cornish, A. J.; Hegg, E. L.; Pachter, R., On understanding proton transfer to the biocatalytic [Fe―Fe]H sub-cluster in [Fe―Fe]H2ases: QM/MM MD simulations. Biochimica et Biophysica Acta (BBA) – Bioenergetics 2011, 1807 (5), 510-517.
4. Lewis, N. S.; Nocera, D. G., Powering the planet: chemical challenges in solar energy utilization. Proceedings of the National Academy of Sciences of the United States of America 2006, 103 (43), 15729-35.
5. Parkin, A., Understanding and Harnessing Hydrogenases, Biological Dihydrogen Catalysts. In The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment, Kroneck, P. M. H.; Torres, M. E. S., Eds. Springer Netherlands: Dordrecht, 2014; pp 99-124.
6. Kates-Harbeck, J., Hydrogen Fuel Cells. Standford University 2010.
7. Cammack, R., Hydrogenase sophistication. Nature 1999, 397 (6716), 214-5.
8. Wait, A. F.; Parkin, A.; Morley, G. M.; dos Santos, L.; Armstrong, F. A., Characteristics of Enzyme-Based Hydrogen Fuel Cells Using an Oxygen-Tolerant Hydrogenase as the Anodic Catalyst. The Journal of Physical Chemistry C 2010, 114 (27), 12003-12009.
9. Ogo, S., H2 and O2 activation by [NiFe]hydrogenases – Insights from model complexes. Coordination Chemistry Reviews 2017, 334, 43-53.
10. Caserta, G.; Roy, S.; Atta, M.; Artero, V.; Fontecave, M., Artificial hydrogenases: Biohybrid and supramolecular systems for catalytic hydrogen production or uptake. Current Opinion in Chemical Biology 2015, 25 (1), 36-47.
11. Caserta, G.; Adamska-Venkatesh, A.; Pecqueur, L.; Atta, M.; Artero, V.; Roy, S.; Reijerse, E.; Lubitz, W.; Fontecave, M., Chemical assembly of multiple metal cofactors: The heterologously expressed multidomain [FeFe]-hydrogenase from Megasphaera elsdenii. Biochimica et Biophysica Acta (BBA) – Bioenergetics 2016, 1857 (11), 1734-1740.
12. Grasse, P. B.; Brauer, B. E.; Zupancic, J. J.; Kaufmann, K. J.; Schuster, G. B., Chemical and physical properties of fluorenylidene: equilibration of the singlet and triplet carbenes. Journal of the American Chemical Society 1983, 105 (23), 6833-6845.
13. Grasa, G. A.; Viciu, M. S.; Huang, J.; Nolan, S. P., Amination Reactions of Aryl Halides with Nitrogen-Containing Reagents Mediated by Palladium/Imidazolium Salt Systems. The Journal of Organic Chemistry 2001, 66 (23), 7729-7737.
14. Budagumpi, S.; Endud, S., Group XII Metal–N-Heterocyclic Carbene Complexes: Synthesis, Structural Diversity, Intramolecular Interactions, and Applications. Organometallics 2013, 32 (6), 1537-1562.
15. Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F., An overview of N-heterocyclic carbenes. Nature 2014, 510 (7506), 485-496.
16. Marynick, D. S., .pi.-Accepting abilities of phosphines in transition-metal complexes. Journal of the American Chemical Society 1984, 106 (14), 4064-4065.
17. Crabtree, R. H., Carbonyls, Phosphine Complexes, and Ligand Substitution Reactions. In The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Inc.: 2005; pp 87-124.
18. Carbonyls, Phosphines, and Substitution. In The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Inc.: 2014; pp 98-133.
19. Pi-Complexes. In The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Inc.: 2014; pp 134-162.
20. Cavell, K., N-Heterocyclic carbenes/imidazolium salts as substrates in catalysis: the catalytic 2-substitution and annulation of heterocyclic compounds. Dalton Transactions 2008, (47), 6676-6685.
21. Hahn, F. E.; Jahnke, M. C., Heterocyclic carbenes: synthesis and coordination chemistry. Angewandte Chemie (International ed. in English) 2008, 47 (17), 3122-72.
22. Lummiss, J. A. M.; Higman, C. S.; Fyson, D. L.; McDonald, R.; Fogg, D. E., The divergent effects of strong NHC donation in catalysis. Chemical Science 2015, 6 (12), 6739-6746.
23. Maeda, Y.; Hashimoto, H.; Kinoshita, I.; Nishioka, T., π-Back-Bonding Interaction Depending on the Bridging Chain Lengths of Chelated N-Heterocyclic Carbene Platinum Units in Heterometallic Trinuclear Complexes Affecting Their Electrochemical Property. Inorganic Chemistry 2014, 53 (2), 661-663.
24. Herrmann, W. A., N-heterocyclic carbenes: a new concept in organometallic catalysis. Angewandte Chemie (International ed. in English) 2002, 41 (8), 1290-309.
25. Li, S.; Jia, W.; Jiao, N., Copper/Iron-Cocatalyzed Highly Selective Tandem Reactions: Efficient Approaches to Z-γ-Alkylidene Lactones. Advanced Synthesis & Catalysis 2009, 351 (4), 569-575.
26. Przyojski, J. A.; Veggeberg, K. P.; Arman, H. D.; Tonzetich, Z. J., Mechanistic Studies of Catalytic Carbon–Carbon Cross-Coupling by Well-Defined Iron NHC Complexes. ACS Catalysis 2015, 5 (10), 5938-5946.
27. Liu, Z.; Derosa, J.; Engle, K. M., Palladium(II)-Catalyzed Regioselective syn-Hydroarylation of Disubstituted Alkynes Using a Removable Directing Group. Journal of the American Chemical Society 2016, 138 (39), 13076-13081.
28. Mercs, L.; Labat, G.; Neels, A.; Ehlers, A.; Albrecht, M., Piano-Stool Iron(II) Complexes as Probes for the Bonding of N-Heterocyclic Carbenes: Indications for π-Acceptor Ability. Organometallics 2006, 25 (23), 5648-5656.
29. Xiang, L.; Xiao, J.; Deng, L., Synthesis, Structure, and Reactivity of Organo-Iron(II) Complexes with N-Heterocyclic Carbene Ligation. Organometallics 2011, 30 (7), 2018-2025.
30. Gao, H.-h.; Yan, C.-h.; Tao, X.-P.; Xia, Y.; Sun, H.-M.; Shen, Q.; Zhang, Y., Synthesis of Anionic Iron(II) Complex Bearing an N-Heterocyclic Carbene Ligand and Its Catalysis for Aryl Grignard Cross-Coupling of Alkyl Halides. Organometallics 2010, 29 (18), 4189-4192.
31. Delville-Desbois, M.-H.; Mross, S.; Astruc, D., Chemistry of (Pentabenzylcyclopentadienyl)iron Compounds Including 17-Electron Dithiocarbamate Complexes. Organometallics 1996, 15 (26), 5598-5604.
32. Wolf, J.; Labande, A.; Daran, J.-C.; Poli, R., Reactivity of Phosphane–Imidazolium Salts Towards [Ir(COD)Cl]2: Preparation of New Hydridoiridium(III) Complexes Bearing Abnormal Carbenes. European Journal of Inorganic Chemistry 2008, 2008 (19), 3024-3030.
33. Jablonskytė, A.; Webster, L. R.; Simmons, T. R.; Wright, J. A.; Pickett, C. J., Electronic Control of the Protonation Rates of Fe–Fe Bonds. Journal of the American Chemical Society 2014, 136 (37), 13038-13044.
34. Capon, J.-F.; El Hassnaoui, S.; Gloaguen, F.; Schollhammer, P.; Talarmin, J., N-Heterocyclic Carbene Ligands as Cyanide Mimics in Diiron Models of the All-Iron Hydrogenase Active Site. Organometallics 2005, 24 (9), 2020-2022.
35. Li, P.; Wang, M.; He, C.; Li, G.; Liu, X.; Chen, C.; Åkermark, B.; Sun, L., Influence of Tertiary Phosphanes on the Coordination Configurations and Electrochemical Properties of Iron Hydrogenase Model Complexes: Crystal Structures of [(μ-S2C3H6)Fe2(CO)6–nLn] (L = PMe2Ph, n = 1, 2; PPh3, P(OEt)3, n = 1). European Journal of Inorganic Chemistry 2005, 2005 (12), 2506-2513.
36. Meyer, J., [FeFe] hydrogenases and their evolution: a genomic perspective. Cellular and molecular life sciences : CMLS 2007, 64 (9), 1063-84.
37. Vignais, P. M.; Colbeau, A., Molecular biology of microbial hydrogenases. Current issues in molecular biology 2004, 6 (2), 159-88.
38. Lamle, S. E.; Albracht, S. P. J.; Armstrong, F. A., The Mechanism of Activation of a [NiFe]-Hydrogenase by Electrons, Hydrogen, and Carbon Monoxide. Journal of the American Chemical Society 2005, 127 (18), 6595-6604.
39. Pandey, A. S.; Harris, T. V.; Giles, L. J.; Peters, J. W.; Szilagyi, R. K., Dithiomethylether as a ligand in the hydrogenase h-cluster. J Am Chem Soc 2008, 130 (13), 4533-40.
40. Peters, J. W.; Schut, G. J.; Boyd, E. S.; Mulder, D. W.; Shepard, E. M.; Broderick, J. B.; King, P. W.; Adams, M. W. W., [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research 2015, 1853 (6), 1350-1369.
41. Reisner, E.; Fontecilla-Camps, J. C.; Armstrong, F. A., Catalytic electrochemistry of a [NiFeSe]-hydrogenase on TiO2 and demonstration of its suitability for visible-light driven H2 production. Chemical Communications 2009, (5), 550-552.
42. Vignais, P. M.; Billoud, B.; Meyer, J., Classification and phylogeny of hydrogenases1. FEMS Microbiology Reviews 2001, 25 (4), 455-501.
43. Volbeda, A.; Charon, M.-H.; Piras, C.; Hatchikian, E. C.; Frey, M.; Fontecilla-Camps, J. C., Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373 (6515), 580-587.
44. Ogata, H.; Lubitz, W.; Higuchi, Y., Structure and function of [NiFe] hydrogenases. Journal of biochemistry 2016, 160 (5), 251-258.
45. Brazzolotto, D.; Gennari, M.; Queyriaux, N.; Simmons, T. R.; Pécaut, J.; Demeshko, S.; Meyer, F.; Orio, M.; Artero, V.; Duboc, C., Nickel-centred proton reduction catalysis in a model of [NiFe] hydrogenase. Nat Chem 2016, 8 (11), 1054-1060.
46. Chen, D.; Ahrens-Botzong, A.; Schünemann, V.; Scopelliti, R.; Hu, X., Synthesis and Characterization of a Series of Model Complexes of the Active Site of [Fe]-Hydrogenase (Hmd). Inorganic Chemistry 2011, 50 (11), 5249-5257.
47. Salomone-Stagni, M.; Stellato, F.; Whaley, C. M.; Vogt, S.; Morante, S.; Shima, S.; Rauchfuss, T. B.; Meyer-Klaucke, W., The iron-site structure of [Fe]-hydrogenase and model systems: an X-ray absorption near edge spectroscopy study. Dalton Transactions 2010, 39 (12), 3057-3064.
48. Xu, T.; Yin, C.-J. M.; Wodrich, M. D.; Mazza, S.; Schultz, K. M.; Scopelliti, R.; Hu, X., A Functional Model of [Fe]-Hydrogenase. Journal of the American Chemical Society 2016, 138 (10), 3270-3273.
49. Shima, S.; Vogt, S.; Göbels, A.; Bill, E., Iron-Chromophore Circular Dichroism of [Fe]-Hydrogenase: The Conformational Change Required for H2 Activation. Angewandte Chemie International Edition 2010, 49 (51), 9917-9921.
50. Jablonskyte, A.; Wright, J. A.; Pickett, C. J., Mechanistic aspects of the protonation of [FeFe]-hydrogenase subsite analogues. Dalton transactions (Cambridge, England : 2003) 2010, 39 (12), 3026-34.
51. Jablonskytė, A.; Wright, J. A.; Fairhurst, S. A.; Webster, L. R.; Pickett, C. J., [FeFe] Hydrogenase: Protonation of {2Fe3S} Systems and Formation of Super-reduced Hydride States. Angewandte Chemie International Edition 2014, 53 (38), 10143-10146.
52. Wang, N.; Wang, M.; Chen, L.; Sun, L., Reactions of [FeFe]-hydrogenase models involving the formation of hydrides related to proton reduction and hydrogen oxidation. Dalton Transactions 2013, 42 (34), 12059-12071.
53. Berggren, G.; Adamska, A.; Lambertz, C.; Simmons, T. R.; Esselborn, J.; Atta, M.; Gambarelli, S.; Mouesca, J. M.; Reijerse, E.; Lubitz, W.; Happe, T.; Artero, V.; Fontecave, M., Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 2013, 499 (7456), 66-69.
54. Esselborn, J.; Lambertz, C.; Adamska-Venkatesh, A.; Simmons, T.; Berggren, G.; Noth, J.; Siebel, J.; Hemschemeier, A.; Artero, V.; Reijerse, E.; Fontecave, M.; Lubitz, W.; Happe, T., Spontaneous activation of [FeFe]-hydrogenases by an inorganic [2Fe] active site mimic. Nat Chem Biol 2013, 9 (10), 607-609.
55. Swanson, K. D.; Ratzloff, M. W.; Mulder, D. W.; Artz, J. H.; Ghose, S.; Hoffman, A.; White, S.; Zadvornyy, O. A.; Broderick, J. B.; Bothner, B.; King, P. W.; Peters, J. W., [FeFe]-Hydrogenase Oxygen Inactivation Is Initiated at the H Cluster 2Fe Subcluster. Journal of the American Chemical Society 2015, 137 (5), 1809-1816.
56. Schwartz, L., Synthetic [FeFe] Hydrogenase Active Site Model Complexes. Universitetsbiblioteket: Uppsala, 2009.
57. Cao, Z.; Hall, M. B., Modeling the Active Sites in Metalloenzymes. 3. Density Functional Calculations on Models for [Fe]-Hydrogenase: Structures and Vibrational Frequencies of the Observed Redox Forms and the Reaction Mechanism at the Diiron Active Center. Journal of the American Chemical Society 2001, 123 (16), 3734-3742.
58. Seyferth, D.; Womack, G. B.; Gallagher, M. K.; Cowie, M.; Hames, B. W.; Fackler, J. P.; Mazany, A. M., Novel anionic rearrangements in hexacarbonyldiiron complexes of chelating organosulfur ligands. Organometallics 1987, 6 (2), 283-294.
59. Hans, M.; Lorkowski, J.; Demonceau, A.; Delaude, L., Efficient synthetic protocols for the preparation of common N-heterocyclic carbene precursors. Beilstein Journal of Organic Chemistry 2015, 11, 2318-2325.
Appendix
Diirion propanedithiolate hexacarbonyl
IR
1,3-dimethylimidazolium iodide 1HNMR
N,N’-bis(2,6-diisopropylphenyl)ethylenediimine 1HNMR
1,3-bis(2,6-diisopropylphenyl)imidazolium chloride 1HMNR
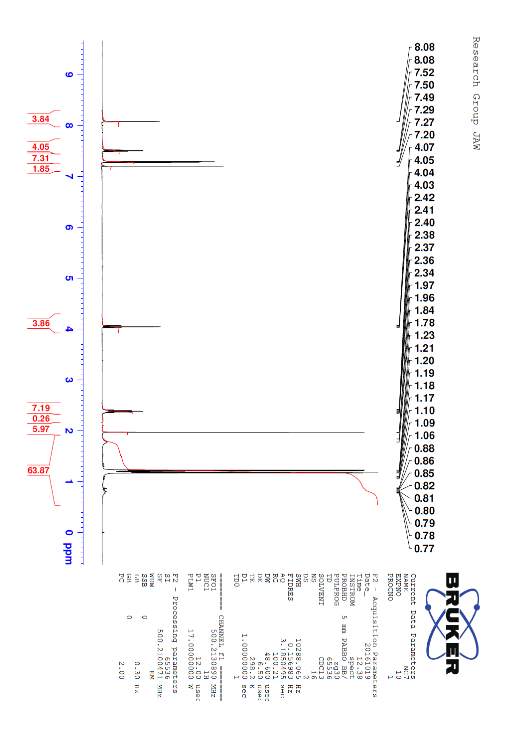
[Fe2(μ-S(CH2)3S)(CO)5(L)] IR Spectra
[Fe2(μ-S(CH2)3S)(CO)5(L)] 13CMNR
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Energy"
Energy regards the power derived from a fuel source such as electricity or gas that can do work such as provide light or heat. Energy sources can be non-renewable such as fossil fuels or nuclear, or renewable such as solar, wind, hydro or geothermal. Renewable energies are also known as green energy with reference to the environmental benefits they provide.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: